Congenital adrenal hyperplasia (CAH)
พญ. รัญชิดา สุวรรณสาร
อาจารย์ที่ปรึกษา ผศ. พญ. ทวิวัน พันธศรี
Congenital adrenal hyperplasia หรือ CAH เป็นโรคที่ถ่ายทอดทางพันธุกรรมจากยีนด้อยบนโครโมโซมร่างกาย (autosomal recessive) ซึ่งเป็นโรคที่เกิดจากการขาดเอนไซม์บางชนิดที่ใช้ในการสังเคราะห์ฮอร์โมนจากต่อมหมวกไต โดยส่งผลให้มีการสร้าง cortisol ที่ลดลงร่างกายจึงตอบสนองด้วยการสร้างฮอร์โมน adrenocorticotropic (ACTH) จากต่อมใต้สมองส่วนหน้า และ corticotropin จาก hypothalamus ที่มากขึ้นจนเกิดเป็น adrenal hyperplasia (1, 2)
การค้นพบ CAH นั้นเกิดขึ้นครั้งแรกในปี 1865 โดยนายแพทย์ชาวอิตาลีนามว่า Luigi De Crecchio จากการชันสูตรศพผู้ป่วยชายที่เสียชีวิตจาก Addisonian crisis ซึ่งผู้ป่วยรายนั้นมีอวัยวะเพศภายนอกเป็นชายแต่มีอวัยวะสืบพันธุ์ภายในเป็นเพศหญิง และมีต่อมหมวกไตขนาดใหญ่ (2) อุบัติการณ์ของ CAH นั้นพบได้น้อยเพียง 1:14,000 ถึง 1:18,000 ต่อการคลอดเท่านั้น (3)
อวัยวะต่างๆของร่างกายสามารถสังเคราะห์ Steroid hormonesชนิดแตกต่างกันได้ เช่น adrenal cortex สร้าง glucocorticoids และ mineralocorticoids, testis สร้าง androgens และ ovaries สร้าง estrogens เป็นต้น โดยอวัยวะที่สำคัญที่จะกล่าวถึงคือ ต่อมหมวกไต (adrenal gland) ซึ่งมี 2 ชั้นได้แก่ adrenal cortex และ adrenal medulla โดย adrenal cortex สามารถแบ่งได้เป็น 3 ชั้น เรียงจากลำดับนอกสุดไปยังในสุดได้แก่
- zona glomerulosa คิดเป็นร้อยละ 15 ของน้ำหนักต่อมหมวกไตทั้งหมด
- zona fasciculata คิดเป็นร้อยละ 75 ของน้ำหนักต่อมหมวกไตทั้งหมด
- zona reticularis คิดเป็นร้อยละ 10 ของน้ำหนักต่อมหมวกไตทั้งหมด (2)
ในแต่ละชั้นทำหน้าที่หลั่งฮอร์โมนที่แตกต่างกัน ดังแสดงในรูปที่ 1
รูปที่ 1 แสดงชั้นต่างๆของต่อมหมวกไต (adrenal gland) และฮอร์โมนที่เกี่ยวข้องของแต่ละชั้น (4)
การสร้างฮอร์โมนจากชั้นต่างๆของ adrenal cortex นั้นต้องอาศัยเอนไซม์หลายชนิด ดังแสดงในรูปที่ 2 จะเห็นได้ว่าเอนไซม์เหล่านี้มีส่วนสำคัญในการสังเคราะห์ steroid hormone ซึ่งในผู้ป่วย CAH จะมีการขาดเอนไซม์ที่แตกต่างกัน ส่งผลให้การสร้างฮอร์โมนเป็นไปอย่างไม่สมบูรณ์ กล่าวคือมีการขาดของฮอร์โมนตัวถัดจากเอนไซม์ตัวที่หายไป (product) และมีการสะสมของฮอร์โมนตัวก่อนหน้า (substrate)
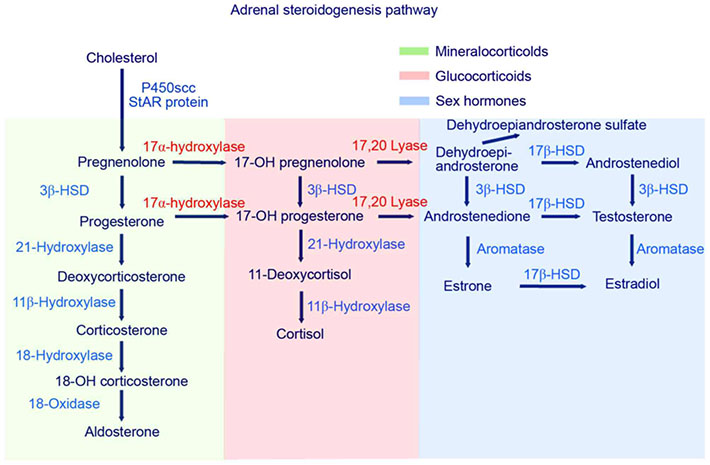
รูปที่ 2 แสดงการสร้าง steroid hormone ชนิดต่างๆจาก adrenal cortex และเอนไซม์ที่เกี่ยวข้อง(5)
CAH สามารถจำแนกได้เป็น 6 ชนิดหลักดังนี้ (2)
1. 21-hydroxylase deficiency (21-OHD)
เป็น CAH ชนิดที่พบบ่อยที่สุด (ร้อยละ 95 ของ CAH ทั้งหมด) (6, 7) และเป็นสาเหตุที่ทำให้ทารกเสียชีวิตจากระบบต่อมไร้ท่อมากที่สุด เกิดจากการขาดเอนไซม์ 21-Hydroxylase หรือ P450c21 ที่ทำหน้าที่เปลี่ยน 17-hydroxyprogesterone (17-OHP) เป็น 11-deoxycortisol และเปลี่ยน progesterone เป็น 11-deoxycorticosterone ซึ่งเป็น pathway ในการสร้าง cortisol และ aldosterone ดังนั้นผู้ป่วยภาวะ 21-OHD จะมีการสะสมของ 17-OHP และมีการสร้าง androgen ที่มากขึ้นเนื่องจากไม่ต้องอาศัย 21-Hydroxylase แต่ทำให้การสร้าง cortisol และ aldosterone ลดลงซึ่งระดับความรุนแรงนั้นสามารถแบ่งได้ดังนี้
- Classical form “Salt-wasting”: ผู้ป่วยมีการขาด cortisol และ aldosterone ในระดับรุนแรงพบได้ประมาณร้อยละ 75 ของ classical CAH ทั้งหมด เกิดจากการ mutation หรือ large deletion ที่รุนแรงของยีน CYP21A2 บนแขนสั้นของโครโมโซมคู่ที่ 6 (6p21.3) จนเหลือการทำงานของ 21-hydroxylase เพียงน้อยกว่าร้อยละ 2 หากไม่ได้รับการรักษาจะทำให้เกิดการขาดน้ำอย่างรุนแรง โซเดียมในเลือดต่ำ โพแทสเซียมในเลือดสูง ระดับฮอร์โมน renin ในเลือดสูง น้ำหนักลด ชัก และเสียชีวิตในช่วง 1 ถึง 4 สัปดาห์หลังคลอดได้ การวินิจฉัยอย่างเร่งด่วนจึงเป็นสิ่งสำคัญ
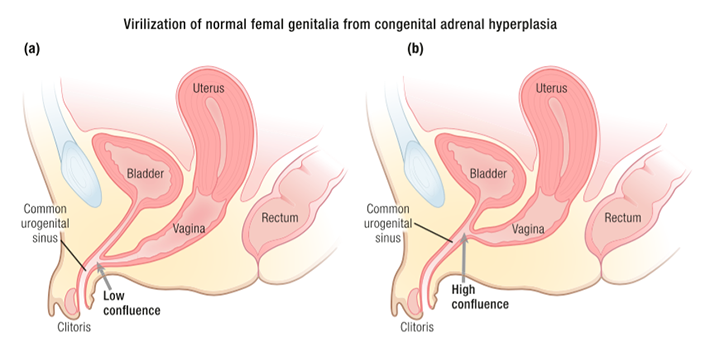
นอกจากนี้ทารกเพศหญิงยังสามารถมีอวัยวะเพศกำกวม (ambiguous genitalia) เช่น enlarged clitoris การเชื่อมกันและมีรอยย่นของ labia majora ท่อปัสสาวะและช่องคลอดไม่แยกจากกัน โดยสาเหตุนั้นมาจากการมีปริมาณ androgen ระดับสูงหากไม่ได้รับการรักษาอาจนำไปสู่การเกิด progressive virilization ได้แก่ การมีขนขึ้นที่หัวหน่าวและรักแร้ก่อนวัย มี advanced skeletal age มีการปิดของ epiphyseal cartilage เร็วกว่าวัยรุ่นทั่วไป - Classical form “Simple virilizing”: คืออีกร้อยละ 25 ที่เหลือของ classical CAH มีการทำงานของ 21-Hydroxylase มากกว่า ชนิด salt-wasting จึงมีความรุนแรงน้อยกว่า สามารถสร้าง aldosterone ที่เพียงพอจนไม่เกิด adrenal crisis หรือ salt loss อย่างไรก็ตามผู้ป่วยในกลุ่ม simple virilizing จะมี androgen สูงและมีอวัยวะเพศกำกวมได้
- Non-classical form: เป็น 21-OHD รูปแบบที่รุนแรงน้อยที่สุด โดยประมาณร้อยละ 80 วินิจฉัยได้ในช่วงอายุ 10 ถึง 40 ปี ซึ่งมีการทำงานของ 21-Hydroxylase ถึงร้อยละ 20-50 แต่ยังมีระดับ cortisol และ aldosterone ที่น้อยกว่าคนปกติจึงทำให้ ACTH อยู่ในระดับที่มากกว่าปกติจนกลายเป็น adrenal hyperplasia และ hyperandrogen อาการที่แสดงได้แก่ การมีขนขึ้นที่หัวหน่าว ก่อนวัย สิวขึ้น ขนดก ประจำเดือนมาผิดปกติ มีบุตรยาก(1, 3, 8)
2. 11β – Hydroxylase deficiency
เป็น CAH ที่พบได้อันดับสอง เกิดจาก mutation ของยีน CYP11B1 ซึ่งอยู่บนแขนยาวของโครโมโซมคู่ที่ 8 (8q21-q22) ทำให้มีการขาดเอนไซม์ 11β – Hydroxylase (P450c11 หรือ CYP11B1) ทำหน้าทีเปลี่ยน 11-deoxycortisol เป็น cortisol และเปลี่ยน 11-deoxycorticosterone (DOC) เป็น corticosterone ซึ่งเป็น precursor ของการสร้าง aldosterone ส่งผลให้มี severe salt-wasting และ simple virilization ได้ อาการที่พบใน 11β-hydroxylase deficiency คือ ความดันโลหิตสูง โพแทสเซียมในเลือดต่ำ ฮอร์โมน renin ในเลือดต่ำ สาเหตุนั้นมาจากการมี DOC ที่สูงขึ้น ซึ่ง DOC นั้นมีฤทธิ์เป็น mineralocorticoid นอกจากนี้ในผู้ป่วยบางรายอาจมีขนดกหรือประจำเดือนมาผิดปกติ (9, 10)
3. 3β – Hydroxysteroid dehydrogenase deficiency (3β-HSD)
เป็นเอนไซม์ที่อยู่ใน glucocorticoid, mineralocorticoid และ androgen pathway ทำหน้าที่เปลี่ยน pregnenolone เป็น progesterone เปลี่ยน 17-Hydroxypregnenolone เป็น 17-Hydroxyprogesterone และเปลี่ยน Dehydroepiandrosterone (DHEA) เป็น androstenedione อาการแสดงของ CAH ชนิดนี้แบ่งได้เป็น salt-wasting และ non-salt-wasting form โดย salt -wasting นั้นสามารถวินิจฉัยได้ในช่วงเดือนแรกๆหลังคลอด ในขณะที่ non-salt-wasting มักเป็นในตอนโต อาจมาด้วยขนหัวหน่าว ขึ้นก่อนวัย หรือ hyperandrogenism (1)
4. 17α – Hydroxylase deficiency
เป็น CAH ที่พบได้น้อยมาก (1 ใน 50,000-100,000) คิดเป็นร้อยละ 1 ของ CAH ทั้งหมด เกิดจากความผิดปกติของยีน CYP17A1 บนโครโมโซมคู่ที่ 10 (10q24.3) ทำหน้าที่เปลี่ยน pregnenolone เป็น 17α-hydroxypregnenolone และเปลี่ยน progesterone เป็น17α-hydroxyprogesterone ผู้ป่วยที่ขาดเอนไซม์นี้จะมี 17α-hydroxypregnenolone, 17α-hydroxylprogesterone ลดลง ส่งผลให้การสร้าง DHEA, androstenedione, testosterone, 11-deoxycortisol, cortisol และ estrogen ลดลงด้วยเช่นกัน ผู้ป่วยหญิงจะมี infantile female genitalia มี underdevelopment of secondary sexual characteristics หรือ อาจะมาด้วย primary หรือ secondary amenorrhea ได้
นอกจากนี้การสะสมของ pregnenolone and progesterone จะไปเพิ่มการสร้าง mineralocorticoid pathway ทำให้มีการเพิ่มขึ้นของ 11-deoxycorticosterone, corticosterone และ18-hydroxycorticosterone ซึ่ง mineralocorticoid เหล่านี้จะทำให้เกิดความดันโลหิตสูงจากการคั่งของน้ำและโซเดียม และมีการสูญเสียโพแทสเซียม แต่จะมีปริมาณ plasma renin ต่ำ(5) อย่างไรก็ตาม ผู้ป่วยจะไม่เกิด adrenal crisis จากการขาด cortisol เนื่องจากมี corticosterone ที่ทำหน้าที่คล้ายกัน (7)
5. P450 oxidoreductase (POR) deficiency
POR เป็นโปรตีนที่เกาะอยู่ที่เยื่อหุ้มเซลล์ ทำหน้าที่ส่งผ่านอิเล็กตรอนให้แก่ P450c17, P450c21, P450aro และเอนไซม์P450s อื่นๆ อาการแสดงนั้นขึ้นกับตำแหน่ง mutation ซึ่งมีความหลากหลายมากในแต่ละประเทศ ความผิดปกติอื่นที่มักพบร่วมด้วยคือ Antley-Bixler syndrome ซึ่งเป็นความผิดปกติของการสร้างกระดูก ได้แก่ craniosynostosis, brachycephaly, radio-ulnar/radio-humeral synostosis, bowed femora, arachnodactyly, midface hypoplasia, proptosis and choanal stenosis เป็นต้น การขาด POR จะทำให้เกิดมี androgen และ cortisol ต่ำ ส่วน mineralocorticoid ปกติ (6, 11)
6. Lipoid congenital adrenal hyperplasia (StAR deficiency)
StAR (steroidogenic acute regulatory protein) ทำหน้าที่นำ cholesterol จาก outer mitochondria membrane เข้ามาสู่ inner mitochondria membrane เพื่อเปลี่ยนเป็น pregnenolone ขั้นตอนนี้ถือเป็น rate limiting step ของกระบวนการสังเคราะห์ steroid hormone ทั้งหมด การเกิด StAR mutation จะทำให้เกิด Lipoid CAH ซึ่งเป็นความบกพร่องในการสร้าง steroid ที่รุนแรงที่สุด ผู้ป่วยจะมาด้วย salt loss, ACTH และ plasma renin สูง รวมไปถึงการตอบสนองต่ำของการสร้าง steroid แม้ถูกกระตุ้นด้วย ACTH นอกจากนี้ยังมีต่อมหมวกไตขนาดใหญ่ที่เต็มไปด้วย cholesterol อาการแสดงนั้นสามารถพบได้ตั้งแต่ช่วงสัปดาห์แรกหลังคลอดไปจนถึงช่วงวัยรุ่นขึ้นอยู่กับความรุนแรงของโรค
กลไกการเกิดคือ ในระยะแรกของ lipoid CAH เมื่อมีการขาด StAR ร่างกายยังสามารถสังเคราะห์ steroid บางส่วนได้จาก StAR-independent pathway แต่เกิดการสะสมของ cholesterol ภายในเซลล์ การสะสมนานเข้าจะสามารถก่อให้เกิดความเสียหายของ mitochondria และเซลล์ ทำให้ไม่สามารถสังเคราะห์ steroid ได้อีกต่อไปดังแสดงในรูปที่ 3 ในผู้ป่วยหญิงจะมีการสร้าง estrogenได้ในช่วง early menstrual cycle แต่จะมีปัญหาในการสร้าง progesterone ใน late menstrual cycle ผู้ป่วยจึงอวัยวะเพศปกติ มีการพัฒนาของเต้านม และมีประจำเดือนจาก estrogen withdrawal bleeding (6, 12)
รูปที่ 3 แสดงถึง lipoid CAH ในระยะต่างๆ(6)
ผู้ป่วย CAH แต่ละชนิดจะมีระดับฮอร์โมนที่ผิดปกติแตกต่างกันออกไป สรุปในตารางที่ 1
|
Enzyme deficiency |
Substrate |
Product |
Mineralocorticoid |
Glucocorticoid |
Androgen |
|
21-Hydroxylase |
Progesterone, 17- OHP |
Deoxycorticosterone, 11-Deoxycortisol |
Decreased |
Decreased |
Increased |
|
11β-Hydroxylase |
Deoxycorticosterone |
Corticosterone |
Increased |
Decreased |
Increased |
|
3β-Hydroxysteroid dehydrogenase |
Pregnenolone, 17-OH pregnenolone, DHEA |
Progesterone, 17-OHP, ADD |
Decreased |
Decreased |
Decreased |
|
17α–Hydroxylase |
Pregnenolone, Progesterone |
17-OH-pregnenolone, 17-OHP |
Increased |
Decreased |
Decreased |
|
P450 oxidoreductase |
Pregnenolone, Progesterone |
DHEA, 11-Deoxycortisol |
Same |
Decreased |
Antenatal increased, Postnatal Decreased |
|
StAR |
Cholesterol |
– |
Decreased |
Decreased |
Decreased |
ตารางที่ 1 สรุปการขาดเอนไซม์ชนิดต่างๆและความผิดปกติของฮอร์โมนในแต่ละชนิด(2)
การซักประวัติและการตรวจร่างกาย
ประวัติที่สำคัญได้แก่ อายุของการมีขนขึ้นที่หัวหน่าว, growth spurt การมีประจำเดือน อาการที่แสดงถึงอาการขาดน้ำ หรือ electrolyte imbalance ต่างๆ อาการคลื่นไส้ อาเจียน อ่อนแรง เกร็ง ชัก ท้องผูก น้ำหนักลด เบื่ออาหาร หน้ามืดเป็นลม รวมไปถึงประวัติครอบครัวที่มีผู้ป่วยเป็น CAH การตรวจร่างกายที่สำคัญได้แก่ การชั่งน้ำหนัก วัดส่วนสูง ความดันโลหิต ดูลักษณะของ hyperpigmentation และ virilization การตรวจ external genitalia อย่างไรก็ตามส่วนมากแล้วอาการแสดงจะหลากหลายและเรื้อรังขึ้นอยู่กับชนิดของ CAH ตามที่กล่าวไปข้างต้น CAH บางชนิดอาจมีอาการที่รุนแรงถึงแก่ชีวิต ดังนั้นแพทย์ควรสามารถแยกความรุนแรงและสามารถส่งตรวจทางห้องปฏิบัติการโดยคร่าวได้ตามตารางที่ 2 (13)
|
|
Adrenal insufficiency |
Adrenal crisis |
|
Symptoms |
Fatigue, Weight loss, Postural dizziness, Anorexia, Abdominal discomfort |
Severe weakness, Syncope, Abdominal pain, Nausea, vomiting, Back pain, Confusion |
|
Signs |
Hyperpigmentation, Low blood pressure, Failure to thrive in children |
Hypotension, Delirium Abdominal tenderness, Reduced consciousness, |
|
Lab results |
Hyponatremia, Hyperkalemia, Hypoglycemia, Hypercalcemia |
Hyponatremia, Hyperkalemia, Hypoglycemia, Hypercalcemia |
ตารางที่ 2 ความแตกต่างของ adrenal insufficiency และ adrenal crisis และการส่งตรวจที่เหมาะสม (13)
การวินิจฉัย
ทำได้โดยการเจาะฮอร์โมนที่จำเพาะกับเอนไซม์ที่ขาดดังนี้
- 21- hydroxylase deficiency: Endocrine Society Clinical Practice Guideline ปี 2018 แนะนำให้เจาะ morning 17 -OHP (ควรตรวจก่อน 8 โมงเช้า ช่วง early follicular phase) ถ้า 17-OHP > 800-1,000 ng/dL (> 24- 30 nmol/L) สามารถวินิจฉัย 21-hydroxylase deficiency ได้เลย และคิดถึงน้อยถ้า 17-OHP < 200 ng/dL (< 6 nmol/L) ในกรณีที่ผลอยู่ระหว่าง 200-800 ng/dL ให้ทำ cosyntropin stimulation test ต่อเพื่อยืนยันการวินิจฉัย ในกรณีที่เป็น non-classical CAH 17-OHP จะมีค่า > 1,000-1,500 ng/dL หลังทำ ACTH stimulation test (1, 3)
- 11β-hydroxylase deficiency จะพบ 11-deoycortisol, 11-deoxycoticosterone และ testosterone สูง
- 3β-Hydroxysteroid dehydrogenase deficiency ให้ทำ ACTH stimulation test จะพบค่า 17α hydroxypregnenolone สูงขึ้น (1)
- 17α-hydroxylase deficiency พบ cortisol, DHEA และ 17OHP ต่ำ แต่ DOC และ corticosterone สูง (7)
การรักษาระหว่างตั้งครรภ์
ในผู้ป่วย CAH ที่ตั้งครรภ์ควรได้รับการตรวจ androstenedione, testosterone และ 17-OHP โดยเพิ่มปริมาณ glucocorticoid ได้เมื่อจำเป็นและควรพิจารณาเลือกเป็น hydrocortisone โดยเฉพาะในช่วงระหว่างรอคลอด ซึ่งพบว่าไม่เพิ่มอัตราการติดเชื้อหรือทำให้แผลหายช้า (1)
การรักษาด้วยการให้ยา
เป้าหมายของการรักษาคือเพื่อป้องกันการเกิด adrenal crisis และ virilization และเพื่อให้ผู้ป่วยมีการเจริญเติบโตรวมไปถึงพัฒนาการที่ปกติ ในเด็กแรกเกิดหรือทารกควรได้ fludrocortisone (mineralocorticoid) และ sodium chloride supplement ร่วมด้วยและอาจให้ปริมาณยาที่มากกว่าปกติ เพื่อช่วยลด adrenal hormone อย่างรวดเร็ว แต่ต้องระมัดระวังไม่ให้เกิด Cushing syndrome ซึ่งเป็นผลจากการรักษา สำหรับในวัยที่ยังเจริญเติบโตอยู่ควรได้ hydrocortisone เนื่องจาก half-life สั้น ทำให้เกิดผลข้างเคียงน้อย มีผลต่อการกดส่วนสูงน้อยกว่าเมื่อเทียบกับ prednisolone และ dexamethasone ในผู้ใหญ่ควรได้รับ hydrocortisone ร่วมกับ fludrocortisone ตามความรุนแรงของโรค (3)
ในผู้ป่วยอายุน้อยกว่าหรือเท่ากับ 18 เดือนควรได้รับการติดตามอย่างใกล้ชิดในช่วง 3 เดือนแรก และควรติดตามต่อทุก 3 เดือน เมื่ออายุมากกว่า 18 เดือนแล้วควรติดตามทุก 4 เดือน ในผู้ป่วยเด็กควรติดตามเรื่องส่วนสูง น้ำหนัก ความดันโลหิต โดยเด็กอายุน้อยกว่า 2 ปีควรตรวจ bone age จนกว่าจะโตเต็มที่ เมื่อเข้าสู่วัยผู้ใหญ่ควรได้รับการวัดความดันโลหิต คำนวณ body mass index และตรวจหาลักษณะของ Cushingoid และเจาะค่าฮอร์โมนเป็นระยะๆ ผู้ป่วยทุกรายควรได้รับการประเมินและเฝ้าระวังอาการแสดงของการขาด mineralocorticoid หรือ glucocorticoid รวมไปถึง androgen excess ด้วย เป้าหมายของการติดตามการรักษาดังสรุปอยู่ในตารางที่ 3 (3)
|
Markers |
Physiology |
Goals |
|
Plasma renin |
Volume status |
ค่าปกติหรือต่ำกว่าปกติ |
|
Potassium |
Mineralocorticoid replacement |
ค่าปกติ |
|
Sodium |
Glucocorticoid and mineralocorticoid replacement |
ค่าปกติ |
|
Testosterone |
Total androgens |
ค่าปกติ |
|
ADD |
Mostly adrenal origin |
ค่าปกติ |
|
Sex hormone- binding globulin |
Testosterone – binding protein |
ใช้คำนวณ free testosterone |
|
17-OHP |
Variable |
ค่าปกติ = overtreatment |
|
Follicular phase progesterone |
Mainly adrenal origin when elevated |
<0.6ng/mL (2nmol/l) |
ตารางที่ 3 การติดตามและเป้าหมายในการรักษาผู้ป่วย CAH (3)
ในกรณีที่มี stress บางอย่าง เช่น อุณหภูมิร่างกายมากกว่า 38.5°C ท้องเสียรุนแรงจนเกิดภาวะขาดน้ำ การออกกำลังกาย มีการผ่าตัดใหญ่หรือได้รับอุบัติเหตุรุนแรงต้องเพิ่มปริมาณ glucocorticoid (3) นอกจากนี้มีการค้นพบว่า ในผู้ป่วย CAH ที่ได้รับการรักษาด้วย glucocorticoid อาจส่งผลให้เกิด insulin resistance ความดันโลหิตสูง ทั้ง systolic และ diastolic และมีโอกาสเกิดการหนาตัวของผนังหลอดเลือด carotid มากขึ้น (14)
สำหรับเรื่องการรักษา hirsutism ใน CAH สามารถให้ oral contraception ร่วมกับ antiandrogen เช่น cyproterone acetate, spironolactone, finasteride, flutamide โดยยาแต่ละตัวให้ผลไม่แตกต่างกัน ขนาดยาตามแสดงในตารางที่ 4 (15)
|
Anti-androgens |
Dosing |
|
Cyproterone acetate |
Day 5 -15: 50-100 mg/day, Day 5 -25: EE 20-35 mg |
|
Spironolactone |
100 – 200 mg/d (divided twice daily) |
|
Finasteride |
2.5 – 5 mg/d |
|
Flutamide |
250 – 500 mg/d (high dose), 62.5 – 250 mg/d (low dose) |
ตารางที่ 4 ปริมาณ antiandrogen ที่ใช้รักษา hirsutism (15)
การรักษาด้วยการผ่าตัด
การผ่าตัดมีเพื่อรักษาอวัยวะเพศกำกวม โดยก่อนผ่าตัดต้องมีการให้ stress dose hydrocortisone ตามตารางข้างต้น นอกจากนี้ยังอาศัย multidisciplinary ได้แก่ กุมารแพทย์ด้านต่อมไร้ท่อ จิตแพทย์ กุมารศัลยแพทย์ ศัลยแพทย์ทางเดินปัสสาวะ โดย position ที่เหมาะสมสำหรับการผ่าตัดคือ lithotomy position (16) การผ่าตัดหลักๆแล้วมีการทำ clitoroplasty หรือ vaginoplasty หรือทั้งสองอย่าง (17)
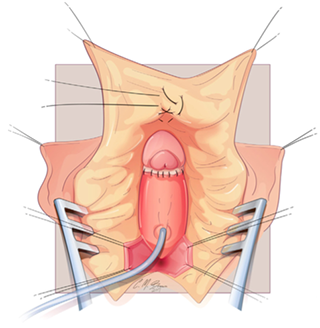
ในกรณีที่ผู้ป่วยมี single urogenital opening แนะนำให้ผ่าตัด urogenital sinus repairing โดยเร็ว (ไม่มีอายุกำหนดที่แน่นอน (3, 16, 18) ดังแสดงในรูปที่ 4
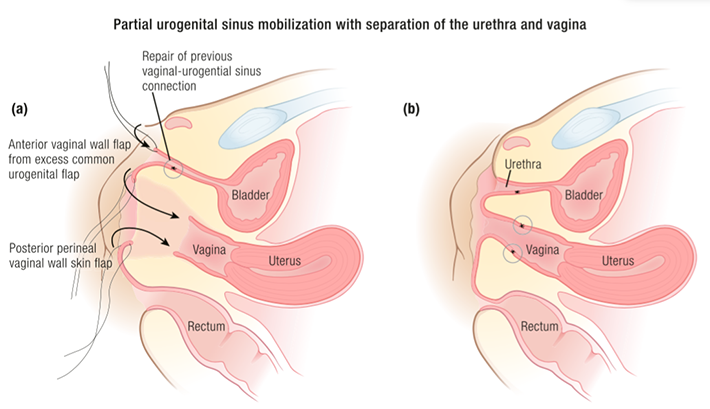
รูปที่ 4 single urogenital opening (ซ้าย) การทำ urogenital sinus repairing (ขวา)(3)
การทำ Clitoroplasty ควรระวัง neurovascular bundle injury ที่อยู่ด้าน dorsal โดยมีการประยุกต์วิธีของ Kogan ซึ่งทำ subtunical reduction clitoroplasty ซึ่งจะเก็บ tunica albugenia ไว้ทั้งหมดเพื่อลดการเกิด glans atrophy และไม่เกิด neurovascular bundle injury ขั้นตอนเริ่มจากการผูกไหมที่ glans clitoris และลงมีดตามแนวรูป A ต่อมาให้ลอก clitoris ผูก tourniquet ที่ base ของ clitoris และลงมีดด้าน ventral ทั้ง 2 ด้านยาวออกให้เห็น corpora carvenosa ที่ถูกปอกออกมาจาก tunica albugenia (รูป B,C,D) ขั้นตอนต่อมาให้ผูกและตัดerectile tissue ด้าน proximal และผูก tunica albuginea ด้าน dorsal และ ventral เข้าด้วยกัน ด้วยไหม 5-0 polydioxanone (รูป E) ผูก glans เข้ากับด้าน ventral ของ tunica albuginea (รูป F) (16) ต่อมาทำ Vaginoplasty โดยการผ่าด้าน dorsal ของ clitoris เป็นแนวยาวเพื่อไปสร้างเป็น labia minora ส่วน clitoral hood ให้สร้างจากหนังด้าน dorsal shaft (16, 19)
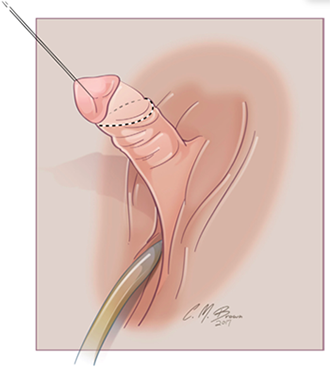
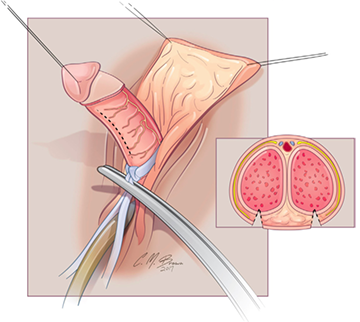
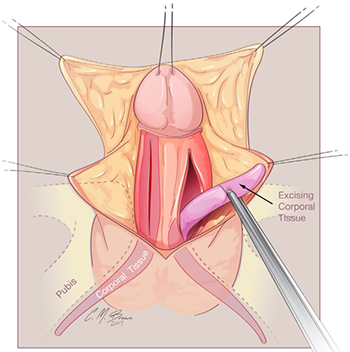
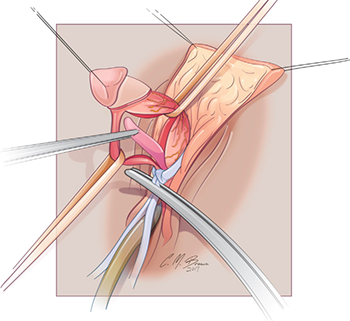
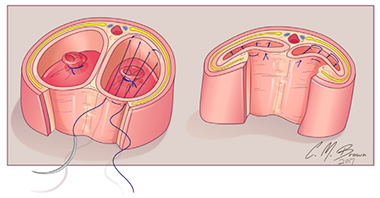
รูปที่ 5 แสดงขั้นตอนการทำ clitoroplasty (16)
มีการรายงานว่าปัญหาที่พบได้หลังการผ่าตัดได้แก่ ช่องคลอดตีบแคบ ปัสสาวะเล็ด ติดเชื้อในระบบทางเดินปัสสาวะ และ sexual dysfunction (20, 21)
เอกสารอ้างอิง
- Fritz MA, Speroff L. Clinical Gynecologic Endocrinology and Infertility. 8th ed: Lippincott Williams & Wilkins (LWW); 2010.
- Podgórski R, Aebisher D, Stompor M, Podgórska D, Mazur A. Congenital adrenal hyperplasia: clinical symptoms and diagnostic methods. Acta Biochim Pol. 2018;65(1):25-33.
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(11):4043-88.
- CNX Os. The adrenal glands. 2009. Available from: https://courses.lumenlearning.com/ap2/chapter/the-adrenal-glands/
- Simiao X, Shuhong H, Xuefeng Y, Muxun Z, Yan Y. 17α‑hydroxylase/17,20‑lyase deficiency in congenital adrenal hyperplasia: A case report. Molecular medicine reports. 2017;15(1):339-44.
- Miller WL. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. Eur J Endocrinol. 2018;179(3):R125-R41.
- Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017;165(Pt A):71-8.
- Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98(7):2645-55.
- Wang D, Wang J, Tong T, Yang Q. Non-classical 11β-hydroxylase deficiency caused by compound heterozygous mutations: a case study and literature review. J Ovarian Res. 2018;11(1):82.
- Kamrath C, Wettstaedt L, Boettcher C, Hartmann MF, Wudy SA. Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 2018;178:221-8.
- Bai Y, Li J, Wang X. Cytochrome P450 oxidoreductase deficiency caused by R457H mutation in POR gene in Chinese: case report and literature review. J Ovarian Res. 2017;10(1):16.
- Bizzarri C, Pisaneschi E, Mucciolo M, Pedicelli S, Galeazzi D, Novelli A, et al. Lipoid congenital adrenal hyperplasia by steroidogenic acute regulatory protein (STAR) gene mutation in an Italian infant: an uncommon cause of adrenal insufficiency. Ital J Pediatr. 2017;43(1):57.
- Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101(2):364-89.
- Tamhane S, Rodriguez-Gutierrez R, Iqbal AM, Prokop LJ, Bancos I, Speiser PW, et al. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2018;103(11):4097-103.
- Martin KA, Anderson RR, Chang RJ, Ehrmann DA, Lobo RA, Murad MH, et al. Evaluation and Treatment of Hirsutism in Premenopausal Women: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(4):1233-57.
- Kaefer M, Rink RC. Treatment of the Enlarged Clitoris. Front Pediatr. 2017;5:125.
- Sturm RM, Durbin-Johnson B, Kurzrock EA. Congenital adrenal hyperplasia: current surgical management at academic medical centers in the United States. J Urol. 2015;193(5 Suppl):1796-801.
- Eckoldt-Wolke F. Timing of surgery for feminizing genitoplasty in patients suffering from congenital adrenal hyperplasia. Endocr Dev. 2014;27:203-9.
- Fernandez-Aristi AR, Taco-Masias AA, Montesinos-Baca L. Case report: Clitoromegaly as a consequence of Congenital Adrenal Hyperplasia. An accurate medical and surgical approach. Urol Case Rep. 2018;18:57-9.
- Almasri J, Zaiem F, Rodriguez-Gutierrez R, Tamhane SU, Iqbal AM, Prokop LJ, et al. Genital Reconstructive Surgery in Females With Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2018;103(11):4089-96.
- Jesus LE. Feminizing genitoplasties: Where are we now? J Pediatr Urol. 2018.
