Common Chromosome Abnormalities
พญ.กฤตยา ภิรมย์
อาจารย์ที่ปรึกษา รศ.ดร.นพ.วีรวิทย์ ปิยะมงคล
ความผิดปกติของโครโมโซม เป็นโรคทางพันธุกรรมที่สำคัญ เนื่องจากทำให้เกิดภาวะแท้งได้ถึง 50 % การตายคลอด 5 % และ สามารถเกิดมามีชีวิตได้ 0.5 % เด็กที่เกิดมาจะพบความผิดปกติ ซึ่งมีผลกระทบต่อชีวิตในระยะยาว การตรวจพบตั้งแต่เบื้องต้นจึงเป็นสิ่งสำคัญ ซึ่งบางภาวะสามารถรักษาได้ ทำให้คุณภาพชีวิตดีขึ้น
การแบ่งกลุ่มความผิดปกติของโครโมโซม
- Numerical abnormalities การผิดปกติที่จำนวนโครโมโซม ซึ่งแบ่งเป็น Polyploidy (จำนวนโครโมโซมเพิ่มขึ้นทั้งชุด) และ Aneuploidy (จํานวนโครโมโซมเพิ่มหรือลดทั้งโครโมโซม)
- Structural abnormalities ได้แก่ Deletions (การหลุดแยกของโครมาทิด), Insertion (การแทรกของโครมาทิด), Inversion (การหมุนสลับตำแหน่งของโครโมโซม), Ring chromosome (โครโมโซมที่มีรูปร่างเหมือนวงแหวน) , Isochromosome (การสลับที่กันของโครโมโซม) และ Translocations(การสับเปลี่ยนชิ้นสวนของโครโมโซม) ซึ่งแบ่งเป็น Reciprocal translocation (Translocation ที่เกิดระหว่างโครโมโซมที่ไม่ใช่ Acrocentric chromosomes 2 โครโมโซม) และ Robertsonian translocation (Translocation ที่เกิดระหว่าง Acrocentric chromosomes 2 โครโมโซม)
Abnormalities of Chromosome Number
Aneuploidy โรคที่มีจํานวนโครโมโซมเพิ่มหรือลดทั้งโครโมโซม ซึ่งในคนปกติมีโครโมโซม 2 ชุด จำนวน 46 แท่ง อาจมีเกินเป็น 47, 48 หรือขาดไปเป็น 44, 45 Trisomy คือโรคที่มีจํานวนโครโมโซมเพิ่มขึ้น 1 โครโมโซม ส่วน Monosomy คือโรคที่มีจํานวนโครโมโซมลดลง 1 โครโมโซม
Autosomal trisomies
Trisomy คือการที่มีโครโมโซมเพิ่มขึ้น 1 โครโมโซม โดยพบประมาณครึ่งหนึ่งของผู้ป่วยที่มีความผิดปกติของโครโมโซม สาเหตุส่วนใหญ่เกิดจากการที่โครโมโซมไม่แยกออกจากกัน หรือ Nondisjunction ซึ่งควรจะแยกออกจากกันในตอนที่มีการแบ่งเซลล์ อาจผิดปกติระยะที่โครโมโซมไม่มาเข้าคู่ หรือการแยกก่อนกำหนด หรือไม่แยก ความเสี่ยงของโรคนี้ขึ้นกับอายุของมารดา (ภาพที่ 1) กล่าวคือมากกว่า 35 ปี และขึ้นกับประวัติการมีลูกเป็น Trisomy โดยโอกาสเสี่ยงที่จะตั้งครรภ์หน้าแล้วมี Trisomy เท่ากับ 1% ดังนั้นจึงควรจะได้รับคำแนะนำในการตรวจวินิจฉัยก่อนคลอดโดยการตรวจโครโมโซมและให้ทางเลือก (1)
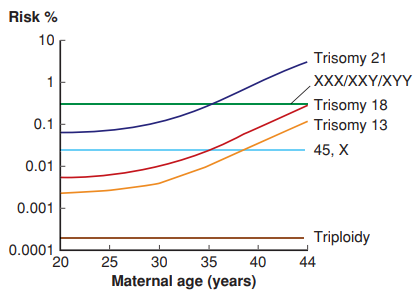
Trisomy 21- Down syndrome
ในปี คศ 1866 กุมารแพทย์ชาวอังกฤษ John Langdon Down ได้ศึกษา กลุ่มเด็กที่บกพร่องด้านสติปัญญา ที่มีลักษณะทางกายภาพที่โดดเด่นและได้อธิบายอาการของดาวน์ไว้ ต่อมาในปี คศ 1959 กุมารแพทย์ชาวฝรั่งเศส Jerome Lejeune อธิบายสาเหตุของโรคว่าเป็นโรคทางพันธุกรรมที่เกิดจากโครโมโซมคู่ที่ 21 เกินมา 1 แท่ง (ภาพที่ 2) (2)

ภาพที่ 2 แสดงkaryotype ของ Down syndrome (47, xy, +21)
สาเหตุ
- เกิดจาก Trisomy 21 คือโครโมโซมคู่ที่ 21 เกินมา 1 แท่ง พบได้ 95% ซึ่งมีสาเหตุจาก Nondisjunction เกิดระหว่าง Meiosis I 75% และ Meiosis II 25%
- เกิดจาก Robertsonian translocation คือมีโครโมโซมย้ายตำแหน่ง พบได้ 3-4 %
- เกิดจาก Isochromosome หรือ Mosaicism คือมีโครโมโซมทั้ง 46 และ 47 แท่ง พบได้ 1 – 2 %
อุบัติการณ์
ดาวน์ซินโดรม เป็นโรคทางพันธุกรรมที่สามารถมีชีวิตอยู่ได้มากที่สุดในกลุ่มโรคโครโมโซมเกิน พบได้ 1 ต่อ 500 ของการตั้งครรภ์ ซึ่งรวมถึงการแท้ง การตายคลอด การเกิดมีชีพ 30 % ของทารกในครรภ์เสียชีวิตตั้งแต่อายุครรภ์ 12 ถึง 40 สัปดาห์ และ 20 % ของทารกในครรภ์เสียชีวิตตั้งแต่อายุครรภ์ 16 ถึง 40 สัปดาห์ ในประเทศสหรัฐอเมริกาพบ โรคดาวน์ซินโดรม 1 ต่อ 740 ของการเกิดมีชีพ หรือ 13.5 ต่อ 10,000 ของการเกิดมีชีพ (3) อุบัติการณ์จะเพิ่มขึ้นตามอายุของมารดา หญิงที่เป็นโรคดาวน์ซินโดรม สามารถมีบุตรได้ และมีโอกาสที่บุตรจะเป็นโรคดาวน์ซินโดรม 1 ใน 3 ส่วนชายที่เป็นโรคดาวน์ซินโดรม จะมีบุตรยากเนื่องจากมีความผิดปกติของการสร้างอสุจิทำให้สร้างได้ปริมาณน้อยมาก (4)
ลักษณะความผิดปกติที่พบได้บ่อย
ประมาณ 25-30% จะพบความผิดปกติตั้งแต่ในครรภ์ โดยการอัลตราซาวน์ที่ไตรมาสที่ 2 ของการตั้งครรภ์ ความผิดปกติของหัวใจ พบได้ประมาณ 40% ของทารกที่เป็นดาวน์ซินโดรม มักเป็นชนิด Endocardial cushion defects และ Ventricular septal defects ความผิดปกติของระบบทางเดินอาหาร พบได้ 7% ได้แก่ ลำไส้ส่วนดูโอดินัมตีบตัน (Duodenal atresia) หลอดอาหารตีบตัน (Esophageal atresia) ลำไส้ใหญ่โป่งพอง (Hirschsprung disease)
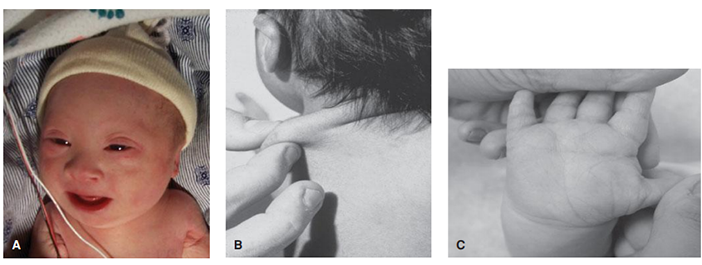
ภาพที่ 3 แสดง A. ลักษณะของดาวน์ซินโดรม B. ผิวที่คอยืด C. เส้นลายมือมักมีเส้นตัดขวางเส้นเดียว
ลักษณะที่พบบ่อย ได้แก่ หัวแบน (Brachycephaly) หัวคิ้วด้านใกล้จมูกทั้ง 2 ข้างมีการหนาตัวขึ้น (Epicanthal folds) มีดวงตาทั้ง 2 ข้างที่เฉียงขึ้นเล็กน้อย (Up-slanting palpebral fissures) ม่านตามีจุดสีขาวที่เรียกว่า Brushfield spots สันจมูกแบน (Flat nasal bridge) กล้ามเนื้อมีความตึงตัวต่ำ (Hypotonia) ทารกมักมีผิวที่คอยืด (Loose skin at the nape of the neck) นิ้วมือสั้น (Short fingers) เส้นลายมือมักมีเส้นตัดขวางเส้นเดียว แทนที่จะแบ่งเป็น 2 เส้น (Single palmar crease) นิ้วก้อยเอียงเข้าหานิ้วนาง (Hypoplasia of the middle phalanx of the fifth finger) มีง่ามนิ้วระหว่างนิ้วโป้งและนิ้วชี้ของเท้ากว้างกว่าปกติ (Sandal-toe gap) (ภาพที่ 3)
ลักษณะของดาวน์ซินโดรมที่กล่าวมาบางอย่างสามารถพบได้จากการอัลตราซาวน์ ดังตารางต่อไปนี้ (1)
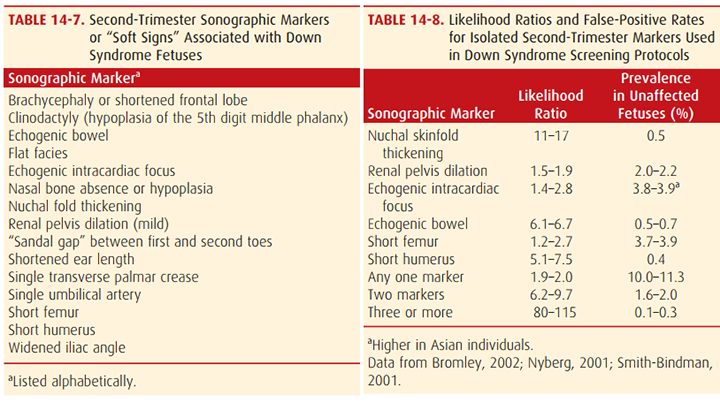
ปัญหาทางสุขภาพที่พบได้ ความผิดปกติทางการได้ยินลดลง 75% มีความผิดปกติทางสายตาส้นหรือยาว 50% ต้อกระจกตั้งแต่แรกเกิด 15% โรคไทรอยด์ 15% มีโอกาสการเกิดมะเร็งเม็ดเลือดขาวสูงขึ้น ระดับของสติปัญญาจะอยู่ในขั้นปัญญาอ่อนเล็กน้อยถึงปานกลาง คือมีไอคิว อยู่ที่ระดับ 35-70 เด็กกลุ่มนี้สามารถอยู่ร่วมกับคนในสังคมได้ดี โดยการเข้าสังคมดีกว่าระดับไอคิว (5)
การพยากรณ์โรค
ทารกที่เป็นดาวน์ซินโดรม สามารถอยู่รอดได้ในปีแรกถึง 95 % อัตรารอดชีวิตใน 10 ปี อย่างน้อย 90% และ 99% ในกลุ่มที่ไม่มีความผิดปกติรุนแรง (6)
Trisomy 18- Edwards syndrome
ในปี คศ 1960 โรคนี้ได้ถูกค้นพบโดย นักพันธุศาสตร์และนักฟิสิกส์ชาวอังกฤษ John Edwards ซึ่งเป็นโรคทางพันธุกรรมที่มีโครโมโซมคู่ที่ 18 เกินมา 1 แท่ง (1)
อุบัติการณ์
โรคนี้พบได้ 1 ต่อ 2,000 ของการตั้งครรภ์ ซึ่งรวมถึงการแท้ง การตายคลอด การเกิดมีชีพ และ 1 ต่อ 6,600 ของการเกิดมีชีพ โรคนี้มีอัตราการเสียชีวิตสูง ซึ่ง 85%จะตายระหว่างอายุคนรรภ์ 10 สัปดาห์ จนถึงกำหนดคลอด มากกว่าครึ่งจะเสียชีวิตในสัปดาห์แรกหลังคลอด อัตราการรอดชีวิตในปี ประมาณ 2% โรคนี้พบในเพศหญิงบ่อยกว่าชาย สามในสี่ของโรคนี้เป็นเพศหญิง (3)
สาเหตุ
โรคนี้เกิดจาก Trisomy 18 คือมีโครโมโซม 18 เกินมา 1 แท่ง
ลักษณะความผิดปกติที่พบได้บ่อย
ความผิดปกติของหัวใจพบได้บ่อยถึง 95% โดยเฉพาะภาวะผนังห้องกั้นหัวใจห้องล่างรั่ว(Ventricular septal defects) ความผิดปกติของสมอง ได้แก่ Cerebellar vermian agenesis, Enlarged cisterna magna, Myelomeningocele ความผิดปกติของช่องท้อง ได้แก่ ไส้เลื่อนกระบังลม (Diaphragmatic hernia) ความผิดปกติของผนังหน้าท้องชนิด Omphalocele การไม่มีรูทวาร (Imperforate anus) ความผิดปกติของไต เช่น Horseshoe kidney
ความผิดปกติของใบหน้าและกระดูก ได้แก่ ส่วนท้ายทอยนูน (Prominent occiput) ใบหูเล็ก (Posteriorly rotated and malformed ears) คางเล็ก (Micrognathia) ปากเล็ก (Small mouth) นิ้วเหลื่อมกัน (Clenched hands with overlapping digits) ไม่มีกระดูกแขน (Radial aplasia) เล็บมีขนาดเล็ก (Hypoplastic nails) เท้ามีลักษณะโค้งคล้ายท้องเรือ (Rocker-bottom feet) โรคเท้าปุก (Clubbed feet) (ภาพที่ 4)
อัลตราซาวน์จะเห็นลักษณะ “Strawberry-shaped” cranium และ Choroid plexus cysts ในการตั้งครรภ์ที่มีความเสี่ยงต่ำ ความเสี่ยงของ Trisomy 18 จะเพิ่มขึ้นก็ต่อเมื่อพบ Choroid plexus cyst ร่วมกับความผิดปกติอื่นๆ การที่พบแค่ Cyst สามารถเป็น ความแตกต่างที่พบ ได้ในคนทั่วไป (ภาพที่ 4)
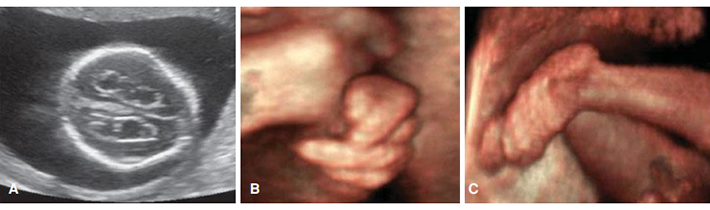
การตั้งครรภ์ในไตรมาสที่สาม มักจะมีภาวะทารกโตช้าในครรภ์ น้ำหนักแรกคลอดโดยเฉลี่ยอยู่ที่ 2,500 กรัม ส่วนวิธีการคลอดควรจะวางแผนไว้ก่อนเพราะระหว่างคลอดมักจะมีการเต้นของหัวใจที่ผิดปกติ สมัยก่อน มากกว่าครึ่งคลอดโดยการผ่าคลอดเนื่องจากทารกอยู่ในภาวะเครียด (Fetal distress) (7)
Trisomy 13 – Patau syndrome
ในปี คศ 1960 นักพันธุศาสตร์ชาวอเมริกัน Klaus Patau และคณะ ได้ รายงานลักษณะความผิดปกติของโรคนี้ไว้และอธิบายว่าเป็นโรคทางพันธุกรรมทีมีโครโมโซมคู่ที่ 13 เกินมา 1 แท่ง (1)
อุบัติการณ์
โรคนี้พบได้ 1 ต่อ 12,000 ของการเกิดมีชีพ และ 1 ต่อ 5,000 ของการตั้งครรภ์ ซึ่งรวมถึงการแท้งและการตายคลอด โรคนี้มีอัตราการเสียชีวิตสูง ซึ่งส่วนใหญ่จะตายระหว่างอายุครรภ์ 10 สัปดาห์ จนถึงกำหนดคลอด (3)
สาเหตุ
- เกิดจาก Trisomy 13 คือมีโครโมโซมเกินมา 1 แท่ง พบได้ 80%
- เกิดจาก Robertsonian translocation พบได้ 20% คือมีโครโมโซมย้ายตำแหน่ง ได้แก่ chromosome 13 และ 14, der(13,14)(q10,q10) ซึ่งเป็น Structural chromosome rearrangement ที่พบมากที่สุด (8)
ลักษณะความผิดปกติที่พบได้บ่อย
ความผิดปกติของสมอง Holoprosencephaly คือการที่สมองส่วนหน้าไม่แยกตัวออกจากกัน พบได้ 2 ใน 3 ของผู้ป่วย และสามารถพบร่วมกับ ภาวะหัวเล็ก (Microcephaly) ภาวะที่ระยะห่างระหว่างเบ้าตาทั้งสองข้างสั้นเข้า (Hypotelorism) และ ความผิดปกติของจมูก คือ มีโพรงจมูกเดียว (Single nostril) และงวง (Proboscis) ความผิดปกติของหัวใจ พบได้ประมาณ 90%ของผู้ป่วย ความผิดปกติของ Neural tube โดยเฉพาะ ภาวะเนื้อสมองหรือเยื่อหุ้มสมองยื่นออกมาจากกะโหลกศีรษะสู่ภายนอก (Cephalocele) ตาเล็ก (Microphthalmia) ปากแหว่งเพดานโหว่ (Cleft lip palate) ความผิดปกติของผนังหน้าท้องชนิด Omphalocele ภาวะถุงน้ำในไต (Cystic renal dysplasia) นิ้วเกิน (Polydactyly) เท้ามีลักษณะโค้งคล้ายท้องเรือ (Rocker-bottom feet) และไม่มีผิวหนัง (Skin aplasia)
ในเด็กที่มี Cephalocele cystic kidneys และ Polydactyly นอกจากจะนึกถึง trisomy 13 แล้ว ยังต้องนึกถึงโรค Meckel-Gruber syndrome ซึ่งเป็นโรคทางพันธุกรรม ชนิด Autosomal recessive ที่ทำให้เสียชีวิตได้ (9)
การพยากรณ์โรค
อายุขัยของเด็กที่เป็น Patau ลดลงอย่างรวดเร็ว โดยอัตราการมีชีวิต ในสัปดาห์แรกหลังคลอด เท่ากับ 40% และอัตราการมีชีวิตรอดที่ 1 ปี เท่ากับ 3% (10)
มารดาที่ตั้งครรภ์บุตรที่เป็นโรคนี้ จะมีความเสี่ยงต่อการเกิดครรภ์เป็นพิษ 50% เนื่องจากบนโครโมโซม 13 มียีนส์ Soluble forms-like tyrosine kinase-1 หรือ sFlt-1 ซึ่งเป็น Antiangiogenic protein ที่เกี่ยวกับภาวะครรภ์เป็นพิษ (10)
Trisomy 16
โรคนี้เกิดจาก Trisomy 16 เป็น Trisomy ที่ไม่สามารถมีชีวิตอยู่รอดได้ พบว่ามีการแท้งมากที่สุดในไตรมาสที่ 1 พบมากถึง 16 %
สาเหตุ
- Full trisomy16 เกิดจาก มีโครโมโซม 16 เกินมา 1 แท่ง
- Mosaic trisomy 16 เกิดจาก มีโครโมโซม 2 แบบ ทั้งโครโมโซมที่ปกติ และ โครโมโซม 16 เกินมา 1 แท่ง
ลักษณะความผิดปกติที่พบได้บ่อย
- Full trisomy16 แท้งตั้งแต่ไตรมาสแรก
- Mosaic trisomy 16 มารดามีความดันโลหิตสูงขณะตั้งครรภ์ มีภาวะทารกโตช้าในครรภ์ คลอดก่อนกำหนด ความผิดปกติของหัวใจ เช่น ventricular septal defect, atrial septal defect ปอดเล็ก ความผิดปกติของกระดูกแลกล้ามเนื้อ ระดับของสติปัญญาต่ำกว่าปกติ
Sex chromosome abnormalities
45, X – Turner syndrome
เทอร์เนอร์เป็นโรคทางพันธุกรรมชนิด Monosomy โรคเดียวที่สามารถมีชีวิตอยู่ได้ และเป็นโรคที่พบอัตราการแท้งมากสุดในกลุ่ม Aneuploidy โดย 20% จะแท้งในไตรมาสแรกของการตั้งครรภ์ (1)
อุบัติการณ์
โรคนี้พบในเพศหญิง โดยพบได้ 1 ต่อ 5,000 การเกิดมีชีพ หรือ 1 ต่อ 2,500 ของเด็กผู้หญิง (3)
สาเหตุ
เกิดจากการหายไปของโครโมโซม ซึ่ง 80% จากบิดา (11)
ลักษณะความผิดปกติที่พบได้บ่อย
ลักษณะที่แสดงออกของโรค แบ่งได้เป็น 3 กลุ่ม ดังนี้
กลุ่มแรก 98% แท้งในไตรมาสแรกของการตั้งครรภ์
กลุ่มที่สอง ช่วงไตรมาสที่1-2 อัลตราซาวน์จะพบก้อนบริเวณคอทารกขนาดใหญ่ (Large cystic hygromas) และ ภาวะทารกบวมน้ำ (Hydrop) ซึ่งทำให้ทารกจะเสียชีวิตในครรภ์ (ภาพที่ 5)
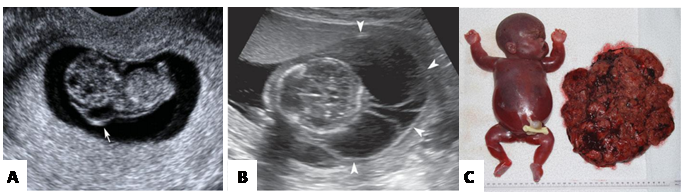
ภาพที่ 5 แสดงถึง Cystic hygroma A. Cyctic hygroma was later found to have Noonan syndrome (1)
B. Massive multiseptated hygroma in hydrops fetalis C. Hydropic, marcerated stillborn infant with large placenta
กลุ่มที่สาม เป็นกลุ่มที่พบน้อยที่สุดและสามารถมีชีวิตอยู่รอด ทารกจะอัลตราซาวน์พบก้อนบริเวณคอขนาดเล็กในไตรมาสที่1-2 และไม่พบภาวะทารกบวมน้ำ ทารกมักพบความผิดปกติที่รุนแรงอื่นๆ
สาเหตุที่พบลักษณะที่แตกต่างกัน เนื่องจาก กลุ่มที่ทารกเกิดมีชีพ จำนวน 1 ใน 2 เป็นชนิด Monosomy และจำนวน 1 ใน 4 เป็น Mosaicism เช่น 45,X/46,XX หรือ 45,X/46,XY ส่วนอีก 15% เป็น Isochromosome X, คือ 46,X,i(Xq) (8)
ลักษณะความผิดปกติอื่นที่พบ ได้แก่ ความผิดปกติของหัวใจ เช่น โรคหลอดเลือดแดงใหญ่คอด (Coarctation of the aorta) ลิ้นหัวใจพิการชนิด bicuspid aortic valve พบได้ 30-50% ความผิดปกติของไต โดยเฉพาะ ไตเป็นรูปเกือกม้า (Horseshoe kidney) ภาวะไทรอยด์ต่ำ ตัวเตี้ย (Short stature) ดังนั้น ในวัยเด็ก มักให้ฮอรโมนการเจริญเติบโต (Growth hormone) เพื่อช่วยเรื่องความสูง หน้าอกกว้าง หัวนมอยู่ห่างกันกว่าปกติ (Broad chest with widely spaced nipples) ภาวะบวมน้ำเหลืองตั้งแต่เกิด (Congenital lymphedema) แผ่นที่คอ (Webbed posterior neck) จาก Cystic hygroma ความผิดปกติของกระดูกและกระดูกอ่อน ระดับของสติปัญญาอยู่ในเกณฑ์ปกติ อาจมีปัญหาความสามารถในการเชื่อมโยงภาพที่เห็นในมิติต่างๆ การใช้ภาษาบกพร่อง การแก้ปัญหาและแปลความหมาย ความผิดปกติในระบบสืบพันธุ์ ผู้ป่วยที่เป็นโรคนี้ 90% มีปัญหารังไข่เจริญน้อย (Ovarian dysgenesis) ทำให้ฮอรโมนเอสโตรเจนต่ำ จึงต้องได้รับฮอรโมนเอสโตรเจนทดแทนก่อนที่จะเข้าวัยรุ่น ในกลุ่มที่เกิดจาก Mosaicism ที่มี โครโมโซมY จะมีความเสี่ยงต่อ ก้อนเนื้องอกที่เกิดจากเซลล์สืบพันธุ์ต้นกำเนิด (Germ cell neoplasm) ซึ่งมีข้อบ่งชี้ในการป้องกันโดยการผ่าตัดต่อมเพศออก (Prophylactic bilateral gonadectomy) (12)
47, XXX – Triple X Syndrome
เป็นโรคทางพันธุกรรมที่เกี่ยวข้องกับโครโมโซมทางเพศ ซึ่งเกิดจากโครโมโซม X เกินมา 1 ตัว
อุบัติการณ์
โรคนี้จะเกิดในผู้หญิง พบได้ประมาณ 1 ในทารกเพศหญิง 1,000 คน โครโมโซม X ที่เกินมา 90% มาจากแม่ (13)
ลักษณะความผิดปกติที่พบได้บ่อย
โรคนี้ไม่ได้พบความผิดปกติชนิดรุนแรง พัฒนาการในวัยรุ่นและการสืบพันธุ์มักปกติ แต่เคยมีรายงานว่ามีภาวะรังไข่หยุดทำงานก่อนกำหนด เป็นหมัน ความสูงอยู่ในเกณฑ์ปกติ ลักษณะที่อาจพบได้ ได้แก่ หัวคิ้วด้านใกล้จมูกทั้ง 2 ข้างมีการหนาตัวขึ้น (Epicanthal folds) นิ้วมือคด (Clinodactyly) กล้ามเนื้อมีความตึงตัวต่ำ (Hypotonia) ไตทำงานผิดปกติ โรคชัก ระดับของสติปัญญาต่ำกว่าปกติ มีสมาธิสั้น พัฒนาการทางภาษาและทักษะการพัฒนากล้ามเนื้อช้า (14)
47, XXY- Klinefelter syndrome
เป็นโรคทางพันธุกรรมที่เกี่ยวข้องกับโครโมโซมทางเพศ ซึ่งเกิดจากโครโมโซม X เกินมา 1 ตัว โดยโครโมโซม X ที่เกินมา มาจากแม่และพ่อเท่ากัน
อุบัติการณ์
เป็นโรคที่พบบ่อยสุดในกลุ่มที่มีความผิดปกติทางด้านโครโมโซมเพศ โรคนี้จะเกิดในผู้ชาย ซึ่งพบได้ 1 ในทารกเพศชาย 600 คน โรคนี้สัมพันธ์กับอายุของแม่และพ่อที่เพิ่มมากขึ้น (13)
ลักษณะความผิดปกติที่พบได้บ่อย
ทารกมีลักษณะเหมือนทารกปกติ และส่วนใหญ่ไม่พบความผิดปกติ ในวัยเด็กมีพัฒนาการปกติ แต่ในวัยรุ่นต่อมเพศจะไม่เจริญ (Gonadal dysgenesis) เป็นหมัน และไม่มีลักษณะแบบเพศชาย คือ ไม่มีหนวดเครา ไม่มีขน อาจมีเต้านม ดังนั้นจึงต้องให้ฮอร์โมนเพศชายเสริมตั้งแต่เริ่มเข้าวัยรุ่น ระดับของสติปัญญาอยู่ในเกณฑ์ปกติ อาจจะต่ำกว่าพี่น้องเดียวกัน การพูด การอ่านและการใช้กล้ามเนื้ออยู่ในเกณฑ์ปกติ (15)
47, XYY
เป็นโรคทางพันธุกรรมที่เกี่ยวข้องกับโครโมโซมทางเพศ ซึ่งเกิดจากโครโมโซม Y เกินมา 1 ตัว
อุบัติการณ์
โรคนี้จะเกิดในผู้ชาย พบได้ 1 ในทารกเพศชาย 1,000 คน โรคนี้ไม่สัมพันธ์กับอายุของพ่อที่เพิ่มมากขึ้น
ลักษณะความผิดปกติที่พบได้บ่อย
ลักษณะที่แสดงออกมาภายนอกปกติ และไม่มีความผิดปกติ ผู้ชายที่เป็นโรคนี้มักจะสูง วัยรุ่นมีพัฒนาการปกติและสืบพันธ์ได้ ระดับของสติปัญญาอยู่ในเกณฑ์ปกติ แต่มีความเสี่ยงที่จะมีปัญหาด้านภาษาทั้งการเขียนและการอ่าน (16)
Abonormalities of chromosome structure
Cri du chat syndrome
เป็นโรคพันธุกรรมชนิดหนึ่งเกิดจากการหลุดหายของส่วนหนึ่งของยีนส์บนแขนข้างสั้นของโครโมโซมคู่ที่ 5 ตำแหน่ง del5p15.2–15.3
อุบัติการณ์
เป็นโรคที่พบบ่อยสุดในกลุ่มที่มีความผิดปกติทางด้าน deletion ซึ่งพบได้ 1 ในทารก 15,000 คน
ลักษณะความผิดปกติที่พบได้บ่อย
มีกล่องเสียงผิดปกติทำให้มีเสียงร้องคล้ายแมว (Cat-like cry) ที่เกิดกับทารกแรกเกิด ศีรษะเล็ก (Microcephaly) หน้ากลม (Round face) จมูกกว้าง (Large nasal bridge) เบ้าตาห่าง (Hypertelorism) หัวคิ้วด้านใกล้จมูกทั้ง 2 ข้างมีการหนาตัวขึ้น (Epicanthal fold) หูเกาะต่ำ (Low-set ears), คางเล็ก (Micrognathia) กล้ามเนื้อมีความตึงตัวต่ำ (Hypotonia) ภาวะสติปัญญาบกพร่อง (17)
Prader willi
เป็นโรคพันธุกรรมชนิดหนึ่งเกิดจากการหลุดหายของส่วนหนึ่งของยีนส์บนแขนข้างยาวของโครโมโซมคู่ที่ 15 ที่ได้รับมาจากพ่อ บนตำแหน่ง 15q11.2–q13 (Paternal genes)
อุบัติการณ์
เป็นโรคที่พบได้ 1 ในทารก 10,000 -30,000 คน
ลักษณะความผิดปกติที่พบได้บ่อย
รูปร่างอ้วน กินจุ เป็นเบาหวาน ตัวเตี้ย ตาเป็นรูปอัลมอนด์ มือเท้าเล็ก กล้ามเนื้อมีความตึงตัวต่ำ (Hypotonia) ภาวะฮอรโมนเพศต่ำ (Hypogonadotropic hypogonadism) หยุดหายใจขณะหลับ (Sleep apnea) พัฒนาการช้า ระดับของสติปัญญาจะอยู่ในขั้นปัญญาอ่อน (18,19)
Angelman
เป็นโรคพันธุกรรมชนิดหนึ่งเกิดจากการหลุดหายของส่วนหนึ่งของยีนส์บนแขนข้างยาวของโครโมโซมคู่ที่ 15 ที่ได้รับมาจากแม่ บนตำแหน่ง 15q11.2–q13 (Maternal genes)
อุบัติการณ์
เป็นโรคที่พบได้ 1 ในทารก 12,000 -20,000 คน
ลักษณะความผิดปกติที่พบได้บ่อย
กลุ่มอาการหุ่นกระบอกที่มีความสุข (“Happy puppet” appearance) ไม่พูด ยิ้มง่าย หัวเราะเก่ง อารมณ์ดี ตื่นเต้นง่าย ชอบปรบมือ ระดับของสติปัญญาจะอยู่ในขั้นปัญญาอ่อน มีการเดินผิดปกติ (ataxia) กล้ามเนื้อมีความตึงตัวต่ำ (Hypotonia) มีอาการชัก (19)
Digeorge syndrome
เป็นโรคทางพันธุกรรมที่เกิดจากการหลุดหายของส่วนหนึ่งของยีนส์บนแขนข้างยาวของโครโมโซมคู่ที่ 22 ตำแหน่ง 22q11.2
อุบัติการณ์
เป็นโรคที่พบได้ 1 ในทารก 2,000 -7,000 คน (1)
ลักษณะความผิดปกติที่พบได้บ่อย
ความผิดปกติของหัวใจ ได้แก่ Conotruncal cardiac defects ปากแหว่งเพดานโหว่ (Cleft palate) การเกิดภาวะความบกพร่องของเพดานอ่อนและผนังคอหอย (Velopharyngeal incompetence) ความผิดปกติของต่อมไทรอยด์และพาราไทรอยด์ (Thymic and parathyroid abnormailites) มีปัญหาด้านการเรียนรู้ (Learning disability) (20)
สรุป
ความผิดปกติของโครโมโซม เกิดจาก 2 สาเหตุ คือ การผิดปกติที่จำนวนโครโมโซม และ การผิดปกติที่รูปร่างโครโมโซม Trisomy ที่พบการแท้งได้มากที่สุดคือ Trisomy 16 และที่สามารถมีชีวิตอยู่ได้มากที่สุดคือ Trisomy 21 ส่วน Turner syndrome เป็นโรคทางพันธุกรรมชนิด Monosomy โรคเดียวที่สามารถมีชีวิตอยู่ได้ และเป็นโรคที่พบอัตราการแท้งมากสุดในกลุ่ม Aneuploidy อีกทั้ง Cri du chat syndromeเป็นโรคที่พบบ่อยสุดในกลุ่มที่มีความผิดปกติทางด้าน deletion
เอกสารอ้างอิง
- Cunningham FG, Leveno KJ, Bloom SL, Spong CY, Dashe JS, Hoffman BL, et al. Williams obstetrics. 24th edition. McGraw-Hill: New York; 2014. p.260-269.
- Kazemi M, Salehi M, Kheirollahi M. Down Syndrome: Current Status, Challenges and Future Perspectives. International Journal of Molecular and Cellular Medicine. 2016;5(3):125-133.
- Dolk H, Loane M, Garne E: The prevalence of congenital anomalies in Europe. Adv Exp Med Biol 686:349, 2010
- Scharrer S, Stengel-Rutkowski S, Rodewald-Rudescu A, et al: Reproduction in a female patient with Down’s syndrome. Case report of a 46,XY child showing slight phenotypical anomalies born to a 47,XX, +21 mother. Humangenetik 26(3):207, 1975
- American Academy of Pediatrics: Health supervision for childen with Down syndromw. Pediatrics 107(2):442, 2001
- Rankin J, Tennant PWG, Bythell M, et al: Predictors of survival in children born with Down syndrome: a registry-based study. Pediatrics 129(6):e1373,2012
- Lin HY, Chen YJ, Hung HY, et al: Clinical characteristics and survival of trisomy 18 in a medical center in Taipei, 1988-2004. Am J Med Genet 140(9):945, 2006
- Nussbaum RL, McInnes RR, Willard HF (eds): Clinical cytogenetics: disorders of the autosomes and sex chromosomes. In Thompson & Thompson Genetics in Medicine, 7th ed. Elsevier-Saunders, 2007, p89
- Lin HY, Lin SP,Chen YJ, et al: Clinical characteristics and survival of trisomy 13 in a medical center in Taiwan, 1985-2004. Pediatr Int 49(3):380, 2007
- Vendola C, Canfield M, Daiger SP, et al: Survival of Texas infants born with trisomies 21, 18, and 13. Am J Med Genet A 152A(2):360, 2010
- Cockwell A, MacKenzie M, Youing S, et al: A cytogenetic and molecular study of a series of 45,X fetuses and their parents. J Med Genet 28(3):151, 1991
- Jones KL: Smith’s Recognizable Patterns of Human Malformation, 6th ed. Philadelphia, Elsevier Saunders, 2006
- Milunsky A, Milunsky JM: Genetic counseling: preconception, prenatal, and perinatal. In Milunsky A (ed): Genetic Disorders of the fetus: diagnosis, Prevention, and Treatment, 5th ed. Baltimore, Johns Hopkins University Press, 2004
- Tartaglia NR, Howell S, Sutherland A, et al: A review of trisomy X (47,XXX). Orphanet J Rare Dis 5:8, 2010
- Girardin CM. Vliet GV: Counseling of a couple faced with a prenatal diagnosis of Klinefelter syndrome. Acta Pediatr 100(6):917, 2011
- Ross JL, Zeger MP, Kushner H, et al: An extra X or Y chromosome: contrasting the cognitive and motor phenotypes in childhood in boys with 47, XYY syndrome or 47,XXY Klienfelter syndrome. Dev Disabil Res Rev 15(4):309,2009
- Cerruti Mainardi P. Cri du Chat syndrome. Orphanet Journal of Rare Diseases. 2006;1:33. doi:10.1186/1750-1172-1-33.
- Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005 Jul 25;7(14):1-20.
- Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010 Aug 15;154C(3):365-76
- Antshel KM, Kates WR, Roizen N, Fremont W, Shprintzen RJ. 22q11.2 deletion syndrome: genetics, neuroanatomy and cognitive/behavioral features keywords. Child Neuropsychol. 2005 Feb;11(1):5-19
