Congenital Adrenal Hyperplasia
อัญญาวีร์ อภิโชติธิวัฒน์
อาจารย์ที่ปรึกษา ร.ศ. นพ. โอภาส เศรษฐบุตร
บทนำ
CAH เป็นโรคทางพันธุกรรมที่ถ่ายทอดทางยีนส์ด้อยเกิดจากการขาดเอ็นไซม์ที่ใช้ในการสังเคราะห์คอร์ติ-ซอลของต่อมหมวกไต โดยจะมีลักษณะอาการของภาวะขาดคอร์ติซอล ร่วมกับภาวะแอนโดรเจนเกิน และอาจมีหรือไม่มีภาวะขาดฮอร์โมนอัลโดสโตโรน ส่วนใหญ่มากกว่า 90% สาเหตุเกิดจากการขาดเอ็นไซม์ 21-hydroxylase
ระบาดวิทยา
โดยในกลุ่ม classic form พบอุบัติการณ์ได้ 1 ใน 15,000 ราย ซึ่งจะแตกต่างไปตามเชื้อชาติและพื้นที่ และในกลุ่ม non-classic form มีอุบัติการณ์ที่สูงกว่ามาก คือ อาจพบได้ 1-2ใน 1000
พยาธิวิทยา
ต่อมหมวกไตมีสองชั้น คือ ชั้นเมดัลลาทำงานสัมพันธ์กับการทำงานของระบบซิมพาเทติกและชั้นคอร์เท็กที่ทำหน้าที่สร้างและหลั่งฮอร์โมนที่เรียกว่า คอร์ติโคสเตียรอยด์ ซึ่งฮอร์โมนกลุ่มนี้สังเคราะห์มาจากสเตียรอยด์คอเลสเตอรอล โดยจะมีโครงสร้างทางเคมีที่คล้ายกัน แต่มีโมเลกุลบางส่วนที่แตกต่างกันเล็กน้อย ทำให้หน้าที่การทำงานต่างกันไป โดยแบ่งได้เป็น 3 ชั้นย่อย คือ
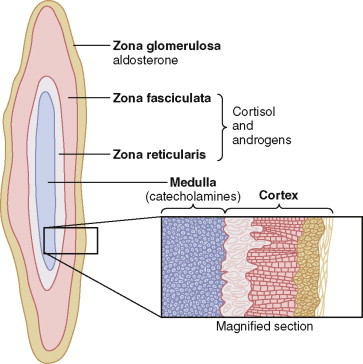
- Zona glomerulosa อยู่ใต้แคปซูล ทำหน้าที่สังเคราะห์อัลโดสเตอโรน
- Zona fasciculata ทำหน้าที่สังเคราะห์ และหลั่งกลูโคคอร์ติคอยด์ฮอร์โมน คอร์ติซอล และ คอร์ติโคสเตอโรน ควบคุมการหลั่งฮอร์โมน เหล่านี้จาก Hypothalamic-Pituitary axis ผ่านทางฮอร์โมน ACTH
- Zona reticularisอยู่ชั้นในสุด ทำหน้าที่สร้าง DHEA และ Androstenedioneซึ่ง sex hormone ที่สร้างออกมามีปริมาณเล็กน้อย แต่ออกฤทธิ์เหมือนเทสโทสเตอโรน
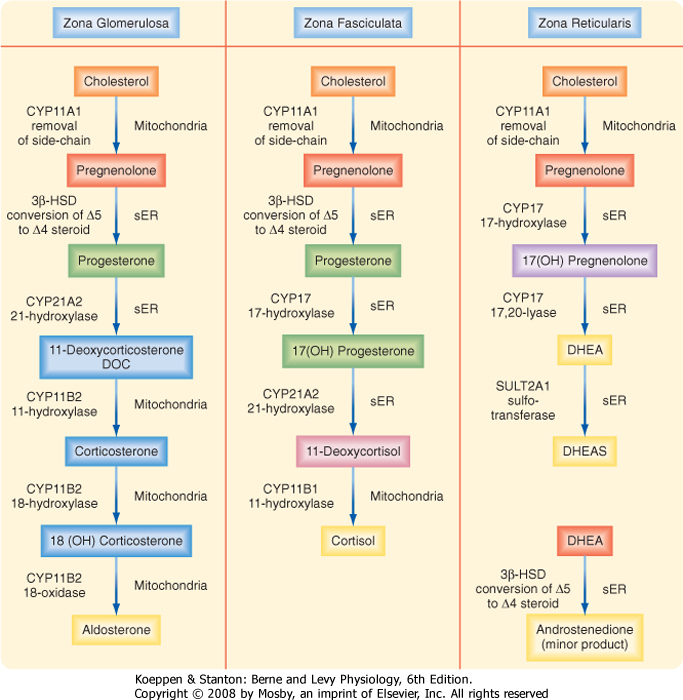
ในภาวะปกติอัลโดสเตอโรนและคอร์ติซอล ถูกควบคุมการสร้างและหลั่งจากกระบวนการที่แตกต่างกัน เช่น Angiotensin เพิ่มปริมาณอัลโดสเตอโรน และทำให้ชั้น zona glomerulosa มีขนาดใหญ่ขึ้นได้ ในขณะที่ ACTH มีผลต่อการสร้างฮอร์โมนคอร์ติซอล และแอนโดรเจน ร่วมกับทำให้ชั้น zona fasciculata และ zona reticularisมีขนาดใหญ่ขึ้นแต่มีผลเพียงเล็กน้อยต่อชั้น zona glomerulosa
ประมาณ 80% ของคอเลสเตอรอลที่ใช้สังเคราะห์สเตียรอยด์ฮอร์โมน มาจากไขมัน LDL ในพลาสมา โดย LDL จะแพร่กระจายจากในแลือดมายัง interstitial fluid และเข้าไปจับกับ ตัวรับเฉพาะ ที่เรียกว่า Coated pits บนผิวของ adrenocortisal cell membranes จากนั้น coated pitsจะถูกนำเข้าเซลล์โดยวิธี Endocytosis สร้างเป็น Vesicle ไปรวมกับ Lysosome เพื่อปล่อยออกมาเป็นคอเลสเตอรอล นำไปสังเคราะห์สเตียรอยด์ฮอร์โมนต่อไป ซึ่งกระบวนการ นำคอเลสเตอรอลเข้าเซลล์นั้น ถูกควบคุมโดย feedback mechanismsเช่น ACTH จะช่วยเพิ่มการสังเคราะห์ฮอร์โมน โดยเพิ่ม LDL receptor มากขึ้น และเร่งการทำงานของเอนไซม์ที่ช่วยเปลี่ยน LDL เป็นคอเลสเตอรอล
เมื่อคอเลสเตอรอลเข้าสู่เซลล์ จะถูกนำไปส่งที่ไมโตคอนเดรีย ซึ่งจะมีเอนไซม์ที่เปลี่ยน คอเลสเตอรอลเป็น Pregnenolone โดยกระบวนการนี้เป็น Rate-limiting step ในการสร้าง adrenal steroid
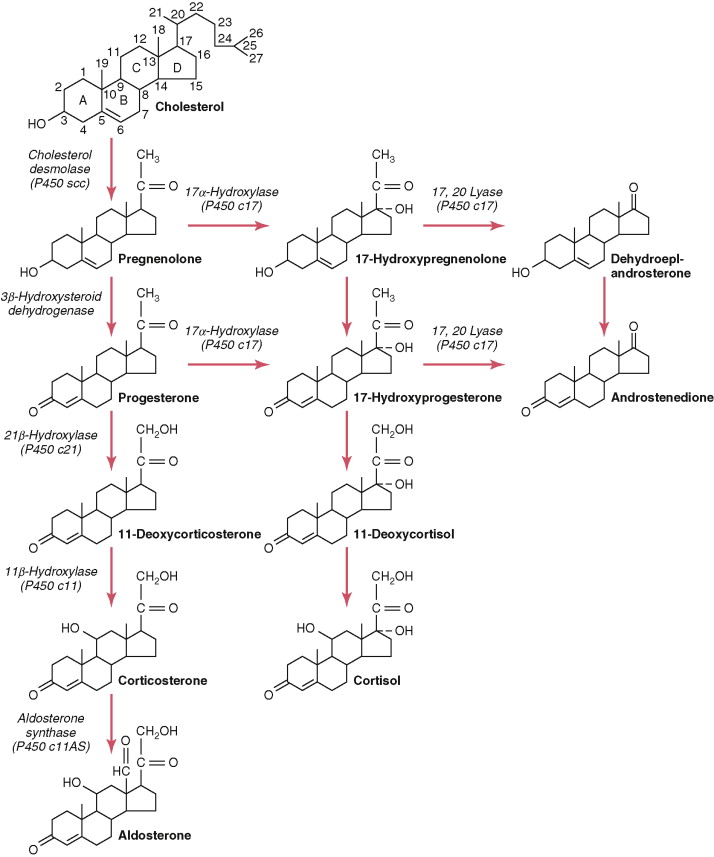
หากมีการเปลี่ยนแปลงของเอนไซม์ตัวใดตัวหนึ่งเพียงเล็กน้อย ก็จะทำให้มีการสร้างที่เปลี่ยนแปลงไปใน pathway ฮอร์โมนอื่นได้
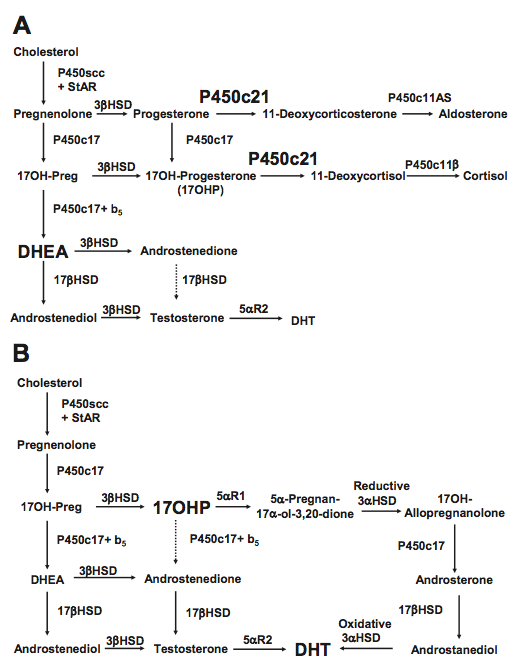
เอนไซม์ 21-hydroxylase (P450c21 หรือ CYP21A2) ทำหน้าที่เปลี่ยน 17-OHP เป็น 11-deoxycortisol (precursor of cortisol) และเปลี่ยนโปรเจสเตอโรน เป็น 11-deoxycorticosterone (precursor of Aldosterone)โดยเมื่อขาดเอ็นไซม์ มีผลทำให้ลดลงของการสร้างคอร์ติซอล เป็น negative feedback ผ่าน HPA axis ส่งผลให้ hypothalamus สร้าง CRH มากขึ้น ไปกระตุ้น pituitary gland มีการหลั่ง ACTH มากขึ้น เกิด Adrenal hyperplasia ( Zona reticularis of adrenal cortex ) และ ทำให้ให้ฮอร์โมนตั้งต้นใน pathway ก่อนการขาดเอ็น-ไซม์มีการสร้างมากขึ้น และไปเพิ่มใน pathway ของการสร้างฮอร์โมนแอนโดรเจน
การที่ร่างกายไม่สามารถสร้างคอร์ติซอลได้ ส่งผลให้สูญเสียเกลือโซเดียม (Salt wasting) มีผลทำให้ผู้ป่วยโตช้า มีภาวะขาดน้ำ และอันตรายถึงช็อคเสียชีวิตได้ และการที่มีแอนโดรเจน ฮอร์โมนที่มาก ในเด็กหญิงก็จะมีอวัยวะเพศกำกวม รวมถึงการเติบโตที่เร็วผิดปกติ นอกจากนี้แล้ว ระดับ CRH ที่สูงเป็นเวลานาน อาจมีผลต่อด้านอารมณ์ ระบบเมตาบอลิซึมพลังงานในร่างกาย เพิ่มโอกาสมีก้อนของต่อมหมวกไตที่ผิดปกติ ในรายที่มีอาการรุนแรงขาดระดับคอร์ติซอลฮอร์โมนมาก จะส่งผลให้การทำงานของ adrenal medulla ผิดปกติไปด้วย มีผลต่อภาวะ stress และการหลั่ง epinephrine ได้
Genetics
CAH มีการถ่ายทอดแบบ autosomal recessive โดยมนุษย์จะมี 2 CYP21A gene อันหนึ่งจะเป็น Nonfunctional pseudogene (CYP21A1 เป็น inactive form ของ enzyme) และอีกอันเป็น active gene ทั้งสองยีนอยู่บนบริเวณเดียวกันของโครโมโซมคู่ที่ 6 (6p21.3) ซึ่งอาจมีการ recombination ได้ในระหว่างกระบวนการ meiosis ทำให้เกิดการ mutation ของ active gene ได้
โดยมีการศึกษาพบว่าหากมี mutation เป็น large deletion หรือ splicing เอนไซม์จะสร้างไม่ได้เลย ซึ่งพบได้ 50% ของ classical CAH, ใน mutationที่มีการเรียงกรดอะมิโนผิดไป เอนไซม์จะพอสร้างได้บ้าง จึงมีลักษณะแสดงออกแบบ simple virillizing, และหากเป็นเพียง point mutation เอนไซม์สามารถทำงานได้ 20–50%ก็จะมีอาการน้อย เป็นเพียง non-classical CAH แต่ความรุนแรงหลากหลายอาจแตกต่างไปได้
ลักษณะอาการทางคลินิก
ความรุนแรงของอาการและอาการแสดงของโรคนั้น ขึ้นกับระดับความรุนแรงของการขาดเอนไซม์ 21-hydroxylase โดยสามารถแบ่งอาการของโรคได้เป็น
- Salt-wasting เป็น classical CAH จะมีอาการที่รุนแรงสุด ขาดทั้งฮอร์โมนคอร์ติซอล และอัลโดสเตอโรน ทำให้มีการเสียทั้งน้ำและเกลือแร่ มีภาวะ Hyponatremia, Hyperkalemia, Acidosis ร่วมกับมีอาการ virilizationมักมีอาการ ให้เห็นตั้งแต่อายุ 2 สัปดาห์แรก
- Simple virilization มีความรุนแรงน้อยกว่า จากการที่ ACTH มีการสร้างที่มากขึ้น สามารถกระตุ้นการสร้าง Mineralocorticoid และ Glucocorticoid ให้เพียงพอได้ แต่แอนโดรเจนที่มากเกินตั้งแต่ในครรภ์ ทำให้มีภาวะ Masculinization ของอวัยวะเพศภายนอก
- Non-classical CAH (late-onest, adult onset adrenal hyperplasia) มีความรุนแรงน้อยสุด จะมีการขาดเอ็นไซม์ไม่มาก พบความผิดปกติได้ในช่วงวัยรุ่น หรือผู้ใหญ่ตอนต้น จากฮอร์โมนแอนโดรเจนที่สูงทำให้มีลักษณะขนที่มาก หรือประจำเดือนผิดปกติได้
Salt-wasting เป็นผลมาจากการขาดฮอร์โมนอัลโดสโตโรน ร่วมกับการที่มี precursor ของ 21-hydroxylase enzyme ที่สูง (17-OHP, Progesterone) ทำหน้าที่เหมือน mineralocorticoid antagonist ทำให้การขาดฮอร์โมนอัลโดสโตโรนรุนแรงมากขึ้น ร่างกายมีการคั่งของโซเดียม สูญเสียน้ำออกมาก มีภาวะขาดน้ำ โพแทสเซียมในร่างกายสูง นอกจากนี้การขาดฮอร์โมนคอร์ติซอลทำให้ร่างกายตอบสนองต่อ Catecholamines ได้ไม่ดี ส่งผลให้มีภาวะช็อคได้ง่ายขึ้น
Ambiguous genitalia โดยทารกที่เป็นโรคจะได้รับฮอร์โมนแอนโดรเจนขนาดสูงตั้งแต่อายุครรภ์ 7สัปดาห์ ทำให้ในเด็กหญิงมีลักษณะอวัยวะเพศกำกวม คือ มีคลิตอริสขนาดใหญ่ บริเวณ labria majara เชื่อมกันและมีรอยพับย่น ช่องเปิดของอวัยวะเพศและทางเดินปัสสาวะเชื่อมรวมกัน แต่พบว่าอวัยวะเพศภายในปกติดี
Postnatal Virilization การที่ได้รับฮอร์โมนเพศขนาดสูงเป็นเวลานาน จะกระตุ้นการเติบโตของร่างกายอย่างรวดเร็ว มีอายุกระดูกที่มากกว่าอายุจริง epiphyseal plate ปิดกอ่นเวลาสมควร มีขนที่อวัยวะเพศและขนรักแร้ขึ้นเร็วกว่าปกติ คลิตอริสมีขนาดโตขึ้น นอกจากนี้ยังไปกระตุ้น HPG axis เกิด precoccoius puberty ได้
ภาวะเจริญพันธุ์ในกลุ่ม classical CAH จะต่ำกว่าผู้หญิงปกติ ประมาณ 60% และในกลุ่ม simple virilization จะประมาณ 80% เนื่องมาจากการไม่ตกไข่เรื้อรังที่สัมพันธ์กับฮอร์โมนแอนโดรเจนที่สูง, ความผิดปกติของอวัยวะเพศภายนอกและปัญหาด้านจิตใจ โดยอัตราการเจริญพันธุ์สัมพันธ์โดยตรงกับความรุนแรงของโรค
ในเพศหญิงที่เป็น classical 21-hydroxylase deficiency ทั้งในกลุ่ม salt-wasting และ simple virilization จะแสดงอาการให้เห็นตั้งแต่แรกเกิด คือ มีอวัยวะเพศภายนอกกำกวม (adrenogenital syndrome) ในเพศชายที่เป็น salt-wasting จะมีอาการในทารกด้วย adrenal insufficiency (เจริญเติบโตช้า ขาดน้ำ hyponatremia hyperkalemia) และในกลุ่ม Simple virilization จะเริ่มมีอาการในวัยเด็กด้วย early virilization
ในเพศหญิงที่เป็น non-classical form หรือ late-onset จะมีอวัยวะเพศภายนอกที่ปกติ แต่จะมาแสดงอาการให้เห็นในภายหลัง คือ ในช่วงเด็กตอนปลายด้วยเรื่องพัฒนาการวัยสาวเร็วกว่าปกติ หรือในช่วงวัยสาว ด้วย
- อาการที่สัมพันธ์กับการตกไข่ที่ผิดปกติ และมีลักษณะคล้าย PCOS (39%)
- อาการขนดกอย่างเดียว โดยไม่มีภาวะประจำเดือนผิดปกติ (39%)
- ไม่มีอาการผิดปกติ แต่มีระดับฮอร์โมนแอนโดรเจนสูงกว่าปกติ (22%)
การวินิจฉัย
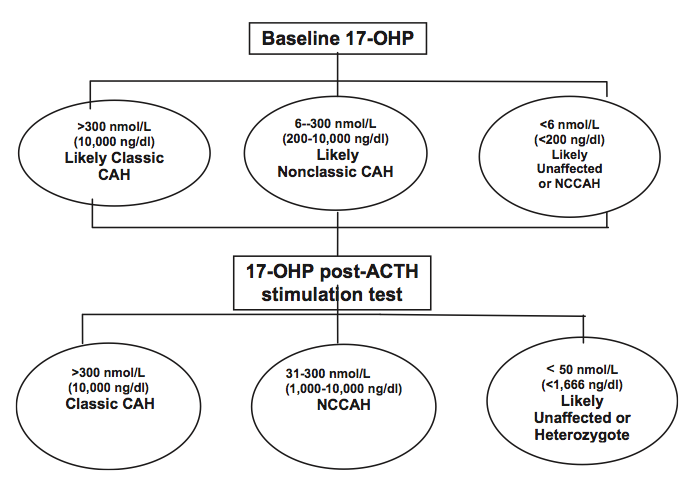
CAH จาก 21-Hydroxylase deficiencyสามารถวินิจฉัยได้จากการที่มีค่า 17-OHP ที่สูงมากกว่าระดับปกติ ตามกลไกของการเกิดโรคดังที่กล่าวมา ในรายที่มีลักษณะการขาดเอนไซม์ที่รุนแรงก็จะมีค่า 17-OHP ที่สูงมากขึ้นตามไปด้วย นอกจากนี้จะมีค่าฮอร์โมนโปรเจสเตอโรนและ androstenedioneสูงตามไปด้วย ในการแยกกับ CAH ที่เกิดจาการขาดเอนไซม์ตัวอื่น อาจพิจารณาดูค่า precusor อื่นเพื่อช่วยแยก
Gold standard ในการวินิจฉัย CAH คือการทำ Corticotropin (cosyntropin) Stimulation Testแล้ววัดระดับ 17-OHP ก่อนและหลังได้ corticotropin 1 ชั่วโมง จะสามารถช่วยวินิจฉัยในรายที่ก้ำกึ่งได้ โดย salt-wasting form จะมีค่าสูงได้ถึง 3000 nmol per literกรณี simple virilizing formจะมีค่าอยู่ในช่วง 300 to 1000 nmol per liter
ในกลุ่ม late-onset nonclassical พบว่า serum 17-OHP จะสูงขึ้นเพียงเล็กน้อย ถ้าค่า morning level <6 nmol per literในช่วง Follicular phase สามารถแยกโรคนี้ออกไปได้ แต่ถ้า>300 nmol per literสามารถวินิจฉัยภาวะนี้ได้ ในคนที่มีค่าก้ำกึ่งต้องทำการตรวจ Corticotorpin stimulation test เพิ่มเติม โดยผู้ป่วยกลุ่มนี้จะมีค่า 17-OHP ที่มากกว่า 50 ถึง 300 nmol per liter
การรักษา
Medical treatment
เป้าหมายในการรักษา เพื่อที่จะลดการหลั่งฮอร์โมนแอนโดรเจนที่มากเกิน ช่วยป้องกันการเกิดภาวะ adrenal crisis และ virilizationรวมถึงมีพัฒนาการการเติบโตที่ปกติ
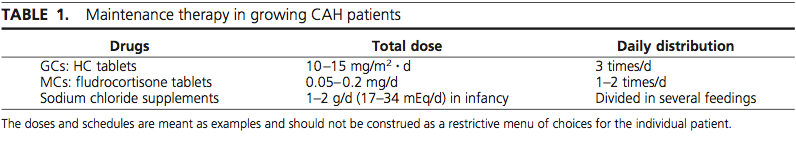
การรักษาจะใช้ Glucocorticoid ในขนาดที่เพียงพอที่จะกดการหลั่งฮอร์โมนแอนโดรเจนของต่อมหมวกไต และเพื่อยับยั้งการหลั่งของ CRH และ ACTH ที่มากเกินไป โดยต้องการใช้ glucocorticoid ในปริมาณที่น้อยที่สุด ที่จะกดการหลั่งฮอร์โมนแอนโดรเจน และทำให้ผู้ป่วยสามารถเติบโตเป็นปกติ และมีน้ำหนักเป็นไปตามเกณฑ์
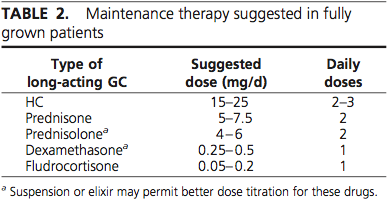
Glucocorticoid ที่เป็นตัวเลือกที่ดีในการรักษา คือ Hydrocortisone โดยเฉพาะในเด็ก ส่วนผู้ใหญ่หรือวัยรุ่นอาจพิจารณาใช้เป็น Dexamethasone หรือ Prednisolone ได้ แต่ยาทุกตัวต้องให้อย่างระมัดระวังการเกิดผลข้างเคียง คือ Cushing’s syndrome
ในรายที่มีปัญหาเรื่อง Salt-wasting การให้ mineralocoticoid เพื่อทดแทนการทำงานของฮอร์โมนอัลโดสเตอโรน นิยมให้เป็น Fludrocortisone และอาจมีการเสริมเกลือโซเดียมด้วย
ในช่วงที่มีภาวะ Stress ต้องทำการเพิ่มขนาดยาเป็น 2-3-เท่าของขนาดปกติและพิจารณาให้ Mineralocorticoid ร่วมด้วย เพื่อทำให้ระดับโซเดียมและโพแทสเซียมปกติผลของการรักษาจะติดตามดูจากระดับ 17-OHP และ Androstenedione รวมถึงเฝ้าระวัง ความแข็งแรงของมวลกระดูก
ผู้ป่วยที่เป็น Non-classic CAH ควรหลีกเลี่ยงการใช้กลูโคคอร์ติคอยในผู้ป่วยที่ไม่มีอาการเพราะ ผลข้างเคียงค่อนข้างสูง แต่ะจะแนะนำในกลุ่มที่มีอาการมาก เช่น เด็กที่มีอาการของ NCCAH ในอายุน้อย (มีการเจริญของขน, bone age ที่สูง) หรือ วัยรุ่นที่มี virilization ค่อนข้างมาก ซึ่งการรักษาจะช่วยลดอาการ hyperandrogenism ได้ ร่วมกับการใช้ oral contraceptive เพื่อรักษาเรื่อง virilization และประจำเดือนที่ผิดปกติ จะพิจารณาหยุดยาได้หากมีอาการที่ดีขึ้น
ในช่วงที่ต้องการตั้งครรภ์ การชักนำการตกไข่อาจมีความจำเป็น
Prenatal diagnosis and treatment
ในมารดาตั้งครรภ์ที่มีความเสี่ยงที่ลูกจะเป็น classic CAHจะพิจารณาให้ Dexamethasone แก่มารดาตั้งแต่ตั้งครรภ์ เพื่อหวังที่จะให้ออกฤทธิ์กด HPA axis ของทารกในครรภ์ ลดการเกิดอวัยวะเพศกำกวม เนื่องจากทารกจะมีการสร้างอวัยวะเพศที่ผิดปกติได้ตั้งแต่ 8 สัปดาห์ หากได้รับฮอร์โมนแอนโดรเจนที่มากเกิน เพราะฉะนั้นควรเริ่มให้ dexamethasone ตั้งแต่ทราบว่าตั้งครรภ์ แต่ทางที่ดีควรมีการตรวจเพศก่อน เพื่อให้ในรายที่ทารกเป็นเพศหญิง โดยพบว่าในทารกเพศหญิงที่เป็น classic CAH หากได้รับยาตลอดการตั้งครรภ์ โอกาสที่เกิดมามีอวัยวะเพศกำกวมหรือผิดปกติเล็กน้อยสูงถึง 85%
แต่Prenantal treatment ยังเป็นข้อโต้แย้ง เนื่องจากโอกาสที่ทารกจะเป็นโรคมีเพียง 1 ใน 8 ในคู่ที่สามีภรรยาเป็นพาหะทำให้มีถึง 7 ใน 8 รายที่ได้รับยาโดยไม่จำเป็น นอกจากนี้การได้รับ dexamethasone ขณะตั้งครรภ์มีโอกาสเกิด Cushing’s syndrome, น้ำหนักขึ้นมากและความดันโลหิตสูงในมารดาได้
Surgical treatment
การผ่าตัดรักษาเพื่อแก้ไขลักษณะอวัยวะเพศกำกวมจะทำในส่วนของ Reconstructive surgery (Clitoplasty, labrioplasty, vaginoplasty)โดยนิยมทำในช่วงที่ผู้ป่วยอายุ 2-6 เดือน เนื่องมาจากช่วงเวลานี้ เนื้อเยื่อมีความยืดหยุ่นได้สูงสุด การผ่าตัดประสบความสำเร็จมากขึ้น อีกทั้งลดการเกิดปัญหาด้านจิตใจของผู้ป่วยด้วย
หากการผ่าตัดไม่ได้ทำในช่วงทารก จะพิจารณาผ่าตัดในช่วงวัยรุ่น กรณีการผ่าตัดแก้ไข ในช่วงอายุที่มากขึ้นมีข้อดี คือ ลดความเสี่ยงที่จะเกิดการตีบตันของช่องคลอด และลดการต้องใช้อุปกรณ์ถ่างขยายช่องคลอดในภายหลัง
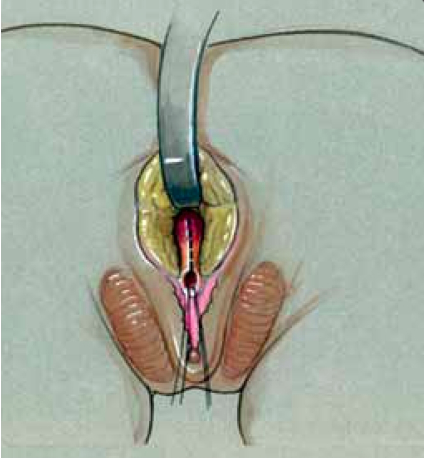
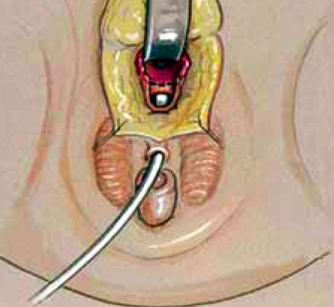
โดยการเทคนิคการผ่าตัดจะป็นอย่างไรนั้น ขึ้นกับความรุนแรงของอวัยวะเพศที่กำกวม และระดับของทางเดินปัสสาวะและช่องคลอดที่มาร่วมกัน ก่อนและหลังการผ่าตัดจะต้องให้ Stress-dose steroid เสมอและทำการเตรียมลำไส้ ผู้ป่วยควรทำ Pelvic Ultrasonography และ Genitography ก่อนผ่าตัด เพื่อประเมินกระเพาะปัสสาวะ ช่องคลอด และ Urogenital sinus (USG) โดยเฉพาะบริเวณที่ช่องคลอดและ USG มาชนกัน เพื่อวางแผนชนิดการผ่าตัด พิจารณาให้ยาปฏิชีวนะเพื่อป้องกันการติดเชื้อก่อนการผ่าตัด
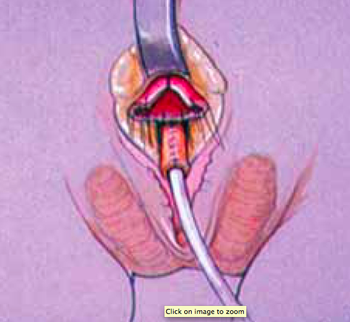
การผ่าตัด นิยมจัดผู้ป่วยในท่า Frog-legged จะเริ่มด้วยการทำ cystovaginoscopy ก่อนเสมอ เพื่อดูเรื่อง anatomyอีกครั้ง และประเมินตำแหน่งของบริเวณที่ช่องคลอดและ USG มาชนกัน เทียบกับ introitus และ bladder neck ข้อดีของการทำ Vaginoplasty พร้อมกับ Labioclitoroplasty คือ สามารถใช้ผิวหนังไปสร้าง labia minora และ vaginoplasty ได้ แต่อาจจำเป็นต้องแก้ไขในบางรายเมื่อเข้าสู่วัยสาว
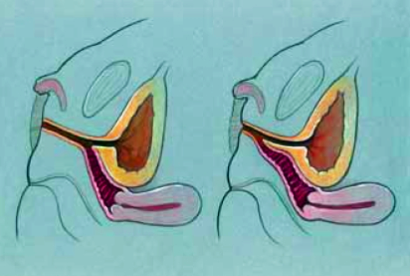
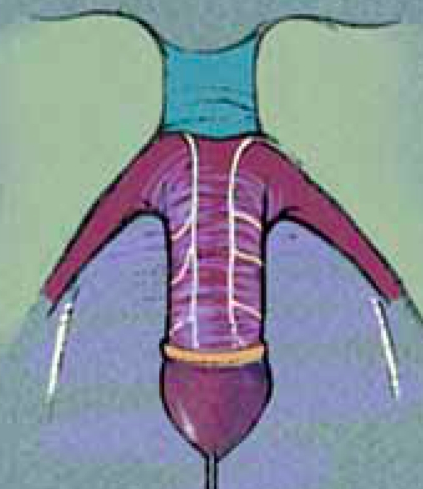
Clitoroplasty คลิตอริสจะมีเส้นเลือดและเส้นประสาทไปเลี้ยงทางด้าน dorsal เพราะฉะนั้นการผ่าตัดต้องระวังในส่วนนี้ในปัจจุบันไม่นิยมทำ clitoral resection เพราะ จะมีอาการปวดมากขณะมี erection โดยหลักการ คือ การเลาะแยก erectile tissue ออกจาก urogenital sinus ก่อน แล้วทำการตัด erectile tissue ทิ้งให้มีส่วน proximal เหลือเล็กน้อย และเก็บ tunica albugina ทางด้าน dorsal เนื่องจากมีเส้นเลือดและเส้นประสาทมาเลี้ยง จากนั้นจึงเย็บส่วนที่ยาวเกินไว้ใต้ pubic bone ส่วนของ glan penis จะนำมาเย็บติด proximal erectile tissue ไว้ เพื่อให้สามารถมีเพศสัมพันธ์ได้ปกติ นำผิวหนังรอบๆมาเย็บเป็น clitoral hood ด้าน dorasl ส่วนด้าน ventral จะรอเย็บหลังทำ vaginoplasty
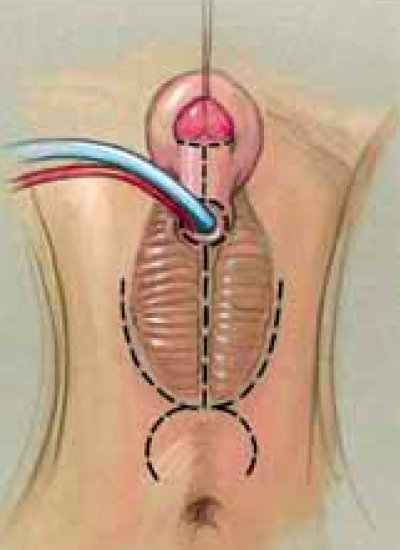
Labioplasty จะทำการตัดแยก prepubial skin ทั้งสองข้างออกเป็น Y shape แล้วดึงมาทางด้าน posterior ข้างต่อ introitus (Y-V advancement) เพื่อทำเป็น Labia majora และเย็บ phallic skin เป็น labia minora
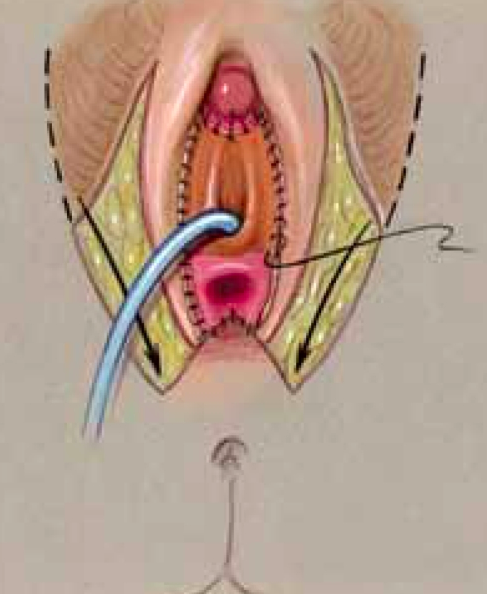
Vaginoplasty ที่ทำในการรักษา CAH จะมี 2 เทคนิคขึ้นกับว่า บริเวณที่มาร่วมกันของ vagina และ urogenital อยู่ระดับไหน โดยแบ่งเป็นposterior skin flapและ pull-throughซึ่งอาจใช้ การผ่าตัด Urogenital mobilization ร่วมด้วย
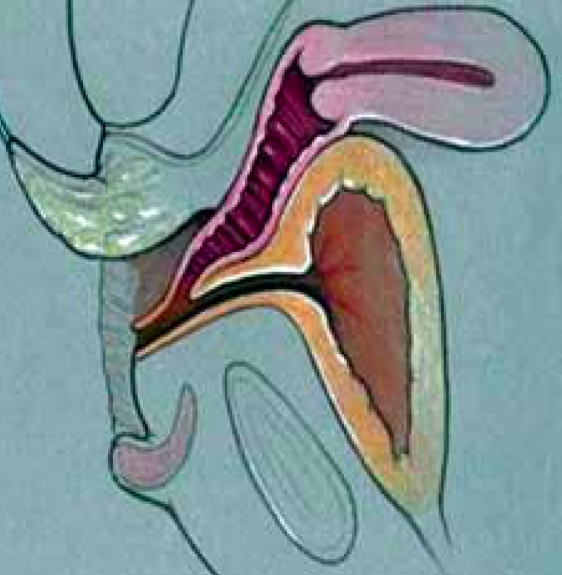
1. Vaginoplasty for low confluence UGS (Flap)
Flap vaginoplasty จะใช้ในกรณีที่มีบริเวณที่มาร่วมกันของ vagina และ urogenital ต่ำหรือปานกลาง เป็นการเลาะเปิดพี้นที่ด้านหลังของ vagina ให้เป็น free space แล้วดึงตัว vagina ให้ลงมาชิด perineum พร้อมกับถ่างขยาย จากนั้นก็เย็บยึดไว้ตรง perineum
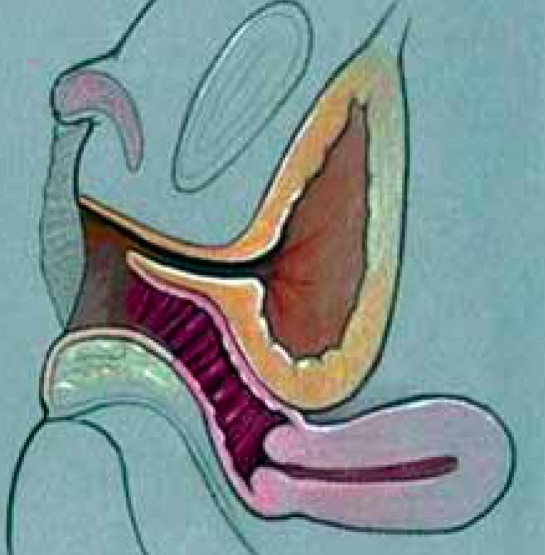
2. Vaginoplasty for moderate or high confluence UGS (Pull-through)
เริ่มจากเลาะ free space หลังต่อ UGS แล้วเปิด tract บริเวณ midline เย็บแยก urethra ก่อน จากนั้นจึงดึงบริเวณ vagina ลงมาใกล้ perineum แล้วทำการเย็บติดกับ skin flap
เอกสารอ้างอิง
- Leslie JA, Cain MP, Rink RC. Feminizing genital reconstruction in congenital adrenal hyperplasia. Indian journal of urology: IJU: journal of the Urological Society of India. 2009;25(1):17.
- Speiser PW, White PC. Congenital adrenal hyperplasia. New England Journal of Medicine. 2003;349(8):776-88.
- Merke DP, Bornstein SR. Congenital adrenal hyperplasia. The Lancet. 2005;365(9477):2125-36.
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism. 2010;95(9):4133-60.
- Hall JE. Guyton and Hall Textbook of Medical Physiology. Philadelphia: Elsevier; 2011.
- Marc A. Fritz LS. Clinical Gynecologic Endocrinology and Infertility. 8 ed. Philadepphia, USA: Lippincott; 2011. 348-59 p.
- Berek JS. Berek&Novak’s Gynecology. 15 ed. Rebar RW, editor. Philadelphia, USA: Lippincott; 2012. 1023-6 p.
- Surgical Management of Congenital Adrenal Hyperplasia [Internet]. Dix D. Poppas. Available from: https://www.cornellurology.com/clinical-conditions/pediatric-urology/genitoplasty-surgery/.
