Array comparative genomic hybridization (aCGH)
พ.ญ. บงกช ชาครบัณฑิต
อาจารย์ที่ปรึกษา ร.ศ. น.พ. โอภาส เศรษฐบุตร
1 Introduction
ความเจริญก้าวหน้าทางพันธุศาสตร์มีมากขึ้นและถูกนำมาใช้ในทางการแพทย์หลายอย่าง ที่นิยมใช้อย่างแพร่หลายในทางสูติศาสตร์ คือการตรวจคัดกรองทารกในครรภ์ (prenatal diagnosis, PND) เพื่อคัดกรองโรครุนแรง เช่นโลหิตจางธาลัสซีเมีย กลุ่มอาการดาวน์ เป็นต้น ในปัจจุบัน มีความเจริญก้าวหน้า ทางเทคโนโลยีมากขึ้น จนสามารถตรวจได้ตั้งแต่ยังไม่มีการตั้งครรภ์ นั่นคือ การตรวจทางพันธุกรรมของ ตัวอ่อนก่อนการตั้งครรภ์ หรือ preimplantation genetic testing ซึ่งมีการตรวจทั้งแบบคัดกรอง (preimplantation genetic screening, PGS) และการตรวจเพื่อการวินิจฉัย (preimplantation genetic diagnosis, PGD)
PGS คือการนำเซลล์ของตัวอ่อนมาใช้ในการตรวจทางพันธุกรรม โดยจุดประสงค์เพื่อการคัดกรองความผิดปกติทางด้านจำนวนโครโมโซมของตัวอ่อน (aneuploidy) ในคู่สมรสที่ปกติ ไม่มีโรคทางพันธุกรรมใดๆ เพื่อหลีกเลี่ยงการย้ายตัวอ่อนที่มีจำนวนโครโมโซมผิดปกติ ทำให้การฝังตัวหลังย้ายตัวอ่อนดีขึ้น(1)
PGD คือการนำเซลล์ของตัวอ่อนมาใช้ในการตรวจทางพันธุกรรม โดยจุดประสงค์เพื่อเลือกตัวอ่อนที่ไม่มีความผิดปกติทางพันธุกรรมในคู่สมรสที่เป็นโรคหรือเป็นพาหะของโรคทางพันธุกกรมใดๆ หรือเพื่อเลือกตัวอ่อนที่มีโครโมโซมที่ต้องการเพื่อจุดประสงค์เฉพาะ เช่น เพศ หรือ แอนติเจนบนเม็ดเลือดขาว เป็นต้น
การตรวจทางพันธุกรรมของตัวอ่อนก่อนการตั้งครรภ์ (preimplantation genetic testing) จำเป็นต้องมีการใช้เทคโนโลยีช่วยในการเจริญพันธุ์ (assisted reproductive technology, ART) เพื่อให้ได้เซลล์มาตรวจก่อนที่จะมีการตั้งครรภ์เกิดขึ้น ซึ่งเซลล์ที่นำมาใช้ในการตรวจทางพันธุกกรมนั้น สามารถได้จากหลายระยะของไข่หรือตัวอ่อน ได้แก่ polar body biopsy, blastomere biopsy และ blastocyst biopsy ซึ่งมีข้อดีข้อเสียแตกต่างกันในแต่ละระยะ
1. Polar body biopsy
Polar body คือเซลล์ที่แบ่งตัวจากไข่ก่อนการปฏิสนธิ สามารถใช้ first หรือ second polar body ก็ได้(2,3) การใช้ polar body สามารถใช้เพื่อตรวจความผิดปกติทางฝั่งมารดาเท่านั้น เนื่องจากยังไม่มีรหัสพันธุกรรม จากฝั่งบิดาโดย polar body จะมีรหัสพันธุกรรมเหมือนไข่หากมารดา มีความผิดปกติชนิด homozygous แต่หากมาดาเป็นพาหะ (heterozygous) polar body จะมีรหัสพันธุกรรม แตกต่างกับไข่ ซึ่งจะสามารถเลือกไข่ที่ปกติเพื่อนำไปปฏิสนธิต่อไป แต่การใช้ polar body biopsy มีข้อจำกัดและข้อเสีย คือ
- หากเกิดการ crossing over ในขณะแบ่งตัวและเป็นตำแหน่งที่เราสนใจ ความแม่นยำในการตรวจ first polar body จะลดลง ดังนั้น จึงควรตรวจ second polar body ด้วย(4)
- การใช้ polar body เป็นการตรวจรหัสพันธุกรรมจากฝั่งมารดาเท่านั้น แม้ว่าจะเป็นไข่ที่มี รหัสพันธุกรรมผิดปกติ แต่หากฝั่งบิดาปกติ ตัวอ่อนที่ได้จะเป็นพาหะ ซึ่งในบางโรค จะไม่แสดงออกทาง phenotype สามารถเติบโตเป็นเด็กปกติได้ ทำให้มีการทิ้งไข่ไปโดยไม่จำเป็น
- เนื่องจากหากมารดามีความผิดปกติแบบ homozygous polar body จะมีรหัสพันธุกรรมเหมือนไข่ จึงไม่จำเป็นต้องตรวจ
2. Blastomere biopsy
ประมาณวันที่ 3 หลังการปฏิสนธิ ตัวอ่อนจะมี 6-8 เซลล์ ยังไม่ยึดกันแน่น (รูปที่ 2) สามารถแยกไปตรวจ ได้ง่าย และยังไม่มีการกำหนดหน้าที่ของแต่ละเซลล์
3. Blastocyst biopsy
ประมาณวันที่ 5-6 หลังการปฏิสนธิ ตัวอ่อนจะอยู่ในระยะ blastocyst จะมีปริมาณเซลล์จำนวนมาก การนำเซลล์มาเพื่อใช้ในการวินิจฉัย จะดึงเซลล์ trophectoderm จากฝั่งที่ไม่มีตัวอ่อน (anembryonic pole) การ biopsy ระยะนี้ทำให้ได้เซลล์ปริมาณมาก ทำให้เพิ่มความแม่นยำในการวินิจฉัย(5) และส่งผลกระทบ กับตัวอ่อนน้อย แต่มีข้อจำกัดในเรื่องการเพาะเลี้ยงเซลล์(6) ซึ่งจะมีตัวอ่อนตายมากขึ้น และเพิ่มโอกาสเกิด monozygotic twin มากขึ้น(7)
Genetic evaluation
1. G-banding karyotyping
การตรวจโครโมโซมด้วย Giemsa banding คือการย้อมสี Giemsa เพื่อให้มองเห็นโครโมโซม โดยการนำเซลล์ที่เพาะเลี้ยงจนอยู่ในระยะ metaphase ย่อยโครโมโซมบางส่วยด้วย Trypsin แล้วนำไปย้อมสี Giemsa การติดสีที่เข้มหรืออ่อน จะแตกต่างกันและเป็นลักษณะจำเพาะของแต่ละยีน ทำให้สามารถตรวจจำนวน และรูปร่างลักษณะของโครโมโซมได้ (8)
2. Fluorescent in situ hybridization (FISH)
เป็นการตรวจหาตำแหน่งที่เฉพาะเจาะจงบนโครโมโซม ในเซลล์ในระยะ interphase หรือ metaphase โดยใช้ RNA หรือ DNA probe ซึ่งเป็น DNA หรือ RNA สายสั้นๆที่ติดสารกัมมันตรังสี ทำปฏิกิริยา hybridization กับ chromosome ที่ต้องการตรวจ จากนั้นนำเซลล์ไปตรวจด้วยกล้องจุลทรรศน์ จะเห็นการเรืองแสงบนโครโมโซมที่จำเพาะต่อ probe ที่ใช้ตรวจ สามารถตรวจได้ครั้งละหลายสี แต่จำนวนสีที่มากขึ้นจะทำให้คุณภาพการอ่านแย่ลง ปัจจุบันที่นิยมตรวจคือ 5 สี ได้แก่การตรวจโครโมโซมคู่ที่ 13, 18, 21, X, Y หรือบางแห่งอาจตรวจ 7 สี โดยเพิ่มโครโมโซมคู่ที่ 16 และ 22 ด้วย
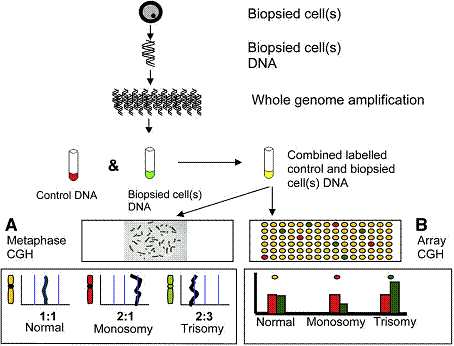
3. Comparative Genomic Hybridization (CGH)(9)
มีการรายงานการใช้ CGH ครั้งแรก ในปี 1992 ในการตรวจหา DNA ของเซลล์มะเร็ง CGH เป็นวิธีที่ใช้ในการตรวจหาการเปลี่ยนแปลงจำนวนของโครโมโซม โดยไม่ต้องเพาะเลี้ยงเซลล์ แต่ใช้การเพิ่มจำนวน DNA ที่ต้องการตรวจ (whole genome amplification, WGA) และติดสารกัมมันตรังสี (fluorochromes) บน DNA ที่ต้องการตรวจ และ DNA ที่ใช้ควบคุม ด้วยสีที่แตกต่างกัน จากนั้นใช้การผสมในอัตราส่วนที่เท่ากัน โดยวิธีที่ใช้ มี 2 วิธี คือ Manual CGH (mCGH) และ array CGH (aCGH)
m-CGH จะใช้ เซลล์ของผู้ชายปกติในระยะ metaphase ยึดบนแผ่นกระจกบาง และทำให้เกิดปฏิกิริยา hybridization ในการตรวจสาย DNA อ่านผลโดยการอ่านสัดส่วนค่าสีจาก probe ตามความยาวของโครโมโซมในระยะนี้ วิธี m-CGH จะใช้เวลาประมาณ 72 ชั่วโมงในการทำปฏิกิริยายังไม่รวมระยะเวลาในการอ่านผล
a-CGH จะใช้ DNA probe ที่เป็นโครโมโซมควบคุม หรือส่วนโครโมโซมที่มีการเพิ่มจำนวนเตรียมไว้ เช่น bacterial artificial chromosome (BAC) oligonucleotide probe ยึดติดบนแผ่น microscope slide แทนการใช้เซลล์ในระยะ metaphase และใช้การเข้าคู่ของโครโมโซมที่ต้องการตรวจ โดยติดด้วยสารกัมมันตรังสีที่แตกต่างกัน จากนั้นใช้การอ่านผลสัดส่วนของสีโดยเครื่องอัตโนมัติ เพื่อดูโครโมโซมที่ขาดหรือเกิน ซึ่งกระบวนการทั้งหมดของ a-CGH จะใช้เวลาได้เร็วที่สุดประมาณ 24 ชั่วโมง ซึ่งหากใช้เซลล์ในวันที่ 3 (blastomere biopsy) จะทำให้สามารถย้ายตัวอ่อนในวันที่ 5 ได้
array ที่นิยมใช้ในปัจจุบัน มี 2 ชนิด คือ targeted arrays โดยการใช้ probe ที่จำเพาะกับโรคหรือความผิดปกติที่ต้องการตรวจ แต่อาจไม่สามารถวินิจฉัยความผิดปกติใหม่ที่เกิดขึ้นได้ และ genome-wide arrays โดยสุ่มตำแหน่งจากโครโมโซมทั้งหมดซึ่งมักใช้ในเชิงศึกษาวิจัยมากกว่า เพราะสามารถค้นพบความผิดปกติ หรือกลุ่มอาการใหม่ๆได้(10)

4. Single nucleotide polymorphism arrays (SNP arrays)(9)
Single nucleotide polymorphism (SNP) คือความผิดปกติของลำดับเบสบน DNA ที่เกิดในประชากรปกติ SNP ส่วนใหญ่เกิดบนทั้ง 2 สายของ DNA เพิ่มจำนวน DNA ด้วย whole genome amplification อาจเลือกเพิ่มจำนวนเป็นบางส่วน หรือเพิ่มทั้ง genome เลยก็ได้ จากนั้นใช้การ hybridization เช่นเดียวกัน สามารถใช้เพื่อแยกโครโมโซมและระบุบุคคลได้ SNP เพิ่งเริ่มนำมาใช้ในการตรวจ PGD จึงยังไม่มีการตีพิมพ์งานวิจัยในเรื่องนี้
Comparative Genomic Hybridization array (a-CGH)
ในการใช้ aCGH มีทั้งข้อดี และข้อเสียแตกต่างกัน โดยเฉพาะเมื่อเทียบกับการใช้วิธี G banding
ข้อดี
1. สามารถวินิจฉัยได้ดีกว่า (higher diagnostic yield)
การตรวจด้วย G banding จะสามารถเห็นความผิดปกติ เมื่อมีขนาด 3-10 ล้าน basepair แต่ความสามารถในการวินิจฉัยของ array จะแตกต่างกันตามชนิดของ array ขึ้นกับขนาดของ probe เช่น oligonucleotide probes มีความละเอียด 50-60 base pairs หรือ BAC มีความละเอียด 50,000 – 200,000 base pair(11) ทำให้การใช้ array สามารถตรวจพบความผิดปกติที่มีขนาดเล็กกว่า การใช้ G banding โดยหากมีความผิดปกติทางโครสร้าง การใช้ a-CGH จะสามารถตรวจพบได้มากกว่า G banding 5.6%(12) พบมีการศึกษาในสตรี 4406 คนซึ่งมารับการตรวจ PND ตามข้อบ่งชี้ต่างๆ เช่น อายุมาก ตรวจคัดกรอง serum marker และอัลตราซาวน์พบความผิดปกติ และตรวจ karyotype พบว่าปกติ เมื่อนำมาตรวจด้วยวิธี aCGH พบว่ามีความผิดปกติที่มีนัยสำคัญทางคลินิก โดยพบร้อยละ 6 ในกลุ่มที่มี structural anomaly และร้อยละ 1.7 ในกลุ่มมารดาอายุมากและพบความผิดปกติจากการตรวจคัดกรอง serum marker(13)
การศึกษาในชิคาโก ปี 2004 โดยการตรวจโครโมโซมของ conceptive product ในผู้ป่วยที่แท้งในไตรมาสแรกของการตั้งครรภ์ที่ผ่านการตรวจด้วยวิธี G-banding มาก่อน พบว่า a-CGH สามารถตรวจพบความผิดปกติได้ทั้งหมด และยังตรวจพบ genomic imbalance อื่นเพิ่มเติมด้วยโดยพบ mosaic ของ trisomy 20 ในชิ้นส่วนรกที่ได้รับการวินิจฉัยเป็น trisomy 21 จาก G-banding พบการเกิด duplication ของ 10q telomere region พบ interstitial deletion ของโครโมโซม 9p และ interstitial duplication ของ Prader-Willi/Angelman syndrome region บนโครโมโซม 15q (14)
2. ใช้เวลาสั้นกว่า (faster turnaround time)
วิธีการตรวจด้วย G banding ต้องมีการเพาะเลี้ยงเซลล์ เพื่อให้ได้ปริมาณ DNA มากพอในระยะที่กำลังแบ่งตัวอย่างเหมาะสม (metaphase) ซึ่งใช้เวลาประมาณ 1-2 สัปดาห์ แต่ในการทำ aCGH สามารถทำได้โดยการสกัด DNA จากเซลล์ได้เลย โดยไม่ต้องผ่านการเพาะเลี้ยง ซึ่งจะสามารถรู้ผลการตรวจได้ภายใน 1 วัน และใช้เครื่องอัตโนมัติในการแปลผล ทำให้มีความแม่นยำ และลดค่าใช้จ่ายจากแรงงานคน
ข้อจำกัด
1. Inability to detect balanced structural rearrangements
ความผิดปกติทางโครงสร้างโครโมโซม จะไม่สามารถตรวจด้วย aCGH ได้ เนื่องจากจำนวนโครโมโซมไม่เปลี่ยนแปลง แต่ความสำคัญทางคลินิกไม่มากนัก เนื่องจาก balance rearrangement พบน้อย โดยพบไม่ถึง ร้อยละ 0.1 ของประชากร (15) มักไม่ก่อให้เกิดโรค หรือความผิดปกติ และหากคู่สมรสมี balance rearrangement ที่ไม่แสดงอาการ บุตรก็จะไม่แสดงอาการเช่นกัน นอกจากนี้ การตรวจพบ balanced rearrangement จากการตรวจ G banding อาจมีความผิดปกติ ด้านจำนวนโครโมโซมใน section ของ DNA ซึ่งไม่สามารถตรวจพบโดย G banding ได้ซ่อนอยู่ พบว่าในกลุ่ม balance translocation มี ความผิดปกติทาง phenotype ประมาณร้อยละ6 (16)
2. Poor ability to detect polyploidies
polyploidies คือการเพิ่มจำนวนโครโมโซมทั้งหมด เช่น 69,XXX เป็นต้น ซึ่งไม่สามารถตรวจได้ ด้วยการทำ standard aCGH เนื่องจาก gene content เปลี่ยนแปลงเท่ากันทั้งหมด ในกรณีที่ทารกเป็น 69,XXY อาจตรวจพบได้เนื่องจาก โครโมโซมเพศไม่เท่ากัน แต่ polyploidies พบน้อยมากในการเก็บข้อมูล จึงยังไม่แสดงความแตกต่างอย่างมีนัยสำคัญทางสถิติ การวินิจฉัย polyploidies มักสงสัยจากการตรวจ พบความผิดปกติจากการตรวจอัลตราซาวน์ตั้งแต่ไตรมาสแรก หรือทารกเสียชีวิตในครรภ์ หรือแรกคลอด ในบางห้องปฏิบัติการณ์จะใช้การตรวจ SNP array หรือใช้ FISH ก่อนการทำ aCGH ซึ่งทำให้สามารถตรวจวินิจฉัย polyploidies ได้ดียิ่งขึ้น (17)
3. Variable ability to detect low level mosaicism
ภาวะ mosaicism ที่เป็นไม่มาก จะให้การวินิจฉัยค่อนข้างยาก และการตรวจ aCGH จะไม่สามารถ mosaism ที่น้อยกว่าร้อยละ 20 ได้ (18)
4. Detection of variants of uncertain significance
variants of uncertain significance คือจำนวนโครโมโซมที่ผิดปกติในตำแหน่งที่ไม่เคยมีรายงาน ว่าควบคุมการแสดงออกของ phenotype อะไร ซึ่งความผิดปกตินี้ตรวจไม่พบโดยวิธี G Banding แต่ตรวจพบจากการตรวจด้วย aCGH ร้อยละ 1-2 (13) มีรายงานข้อมูลในปี 2008-2011 ว่า aCGH ตรวจพบ VUS ประมาณร้อยละ 3.4 ซึ่งตรวจ karyotype ปกติ แต่คาดการณ์ว่าเมื่อความรู้ในสาขานี้พัฒนาขึ้น VUS จะลดลงเนื่องจากเมื่อนำ VUS ดังกล่าวมาวิเคราะห์ใหม่ในปี 2012 พบว่า VUS ลดลงเหลือร้อยละ 1.5 (13) นอกจากนี้ การตรวจพบ VUS ยังก่อให้เกิดคำถามด้านจริยธรรมว่า จะแจ้งผลการตรวจให้คู่สมรส หรือไม่ และคู่สมรสจะตัดสินใจย้ายตัวอ่อนหรือไม่
5. Expense
aCGH มีราคาสูง แต่แนวโน้มราคาค่อยๆลดลง
6. Variable clinical sensitivity
Clinical sensitivity จะสูงหากความผิดปกติทางโครโมโซมนั้นเกิดจากการเพิ่ม หรือลดปริมาณ โครโมโซม (microdeletion หรือ microduplication syndrome) เช่น Velocardiofacial syndrome ซึ่งเกิดจาก 22q11.2 deletion CMA จะ detect ได้ มากกว่าร้อยละ 90 ซึ่งในกรณีที่ตรวจไม่พบ deletion จาก CMA มักเป็นการวินิจฉัยโรคอื่น ที่มีอาการใกล้เคียงกัน แต่หากกลุ่มอาการผิดปกติ เกิดได้จากหลายความผิดปกติทางโครโมโซม เช่น neurofibromatosis type-1 (NF1) ซึ่งประมาณร้อยละ 5 เกิดจาก microdeletion ส่วนใหญ่เกิดจาก ความผิดปกติของลำดับ base ที่ผิดปกติไป ซึ่งกรณีนี้จะไม่สามารถตรวจโดย CMA ได้
Clinical indication
ACOG recommendation (10)
มีหลายๆงานวิจัยที่แสดงให้เห็นว่า aCGH เป็นเครื่องมือที่ดีในการตรวจคัดกรองความผิดปกติขณะตั้งครรภ์ โดยเฉพาะกรณีที่การตรวจ karyotyping ไม่พบความผิดปกติ โดย ACOG ได้ให้คำแนะนำ สำหรับการใช้ aCGH ในการตรวจคัดกรองความผิดปกติขณะตั้งครรภ์ดังนี้ (ACOG committee opinion 2009)
- Conventional karyotyping ยังคงแนะนำให้ใช้เป็นวิธีแรกในการตรวจคัดกรองทารกในครรภ์
- อาจให้คำปรึกษาและให้ทางเลือกคู่สมรส ในการตรวจ targeted aCGH เพิ่มเติม หากตรวจพบ ความผิดปกติจากการอัลตราซาวน์ แต่ตรวจ karyotyping ไม่พบความผิดปกติ หรือในกรณีที่ทารก เสียชีวิตในครรภ์ที่ไม่สามารถเพาะเลี้ยงเซลล์เพื่อตรวจ conventional karyotyping ได้
- คู่สมรสที่เลือกวิธี aCGH ควรได้รับคำแนะนำ ทั้งก่อนและหลังการตรวจ และคู่สมรสควรมีข้อมูลว่า การตรวจ aCGH ไม่สามารถตรวจวิเคราะห์ความผิดปกติทางพันธุกรรมได้ทั้งหมด และบางครั้งอาจแปลผลได้ยาก
- Targeted CGH อาจมีประโยชน์ในการใช้คัดกรองความผิดปกติของโครโมโซม แต่การจะนำมาใช้เป็นวิธีแรกในการตรวจแทน conventional karyotyping ยังต้องรอการศึกษาในอนาคต
ACMG recommendation (11)
นอกเหนือจากทางสูติกรรมที่ใช้ประโยชน์จากการตรวจ aCGH ขณะตั้งครรภ์ ยังมีข้อบ่งใช้ ในการตรวจทางอายุกรรมในทารกหลังคลอดด้วย โดยเฉพาะการเกิดภาวะพัฒนาการช้า (developmental delay, DD) หรือ การเรียนรู้ผิดปกติ (Intellectual disability, ID) American college of Medical Genetics (ACMG) ให้คำแนะนำเกี่ยวกับการใช้ a-CGH หลังคลอด (2010) ดังนี้
- แนะนำให้ใช้ CMA ในการตรวจหา Copy number variants เป็นวิธีการแรกสำหรับ
- สำหรับกรณีทารกมีความผิดปกติหลายอย่างที่ไม่จำเพาะ กับกลุ่มอาการใดๆทางพันธุกรรม
- กรณีมี DD หรือ ID ที่ไม่จำเพาะกับกลุ่มอาการใดๆ
- Autism spectrum disorders
- ใช้ในการตรวจเพิ่มเติมกรณีที่เด็กมีปัญหาเรื่องการเจริญเติบโต (growth retardation) พัฒนาการการพูดช้า หรือภาวะอื่นๆที่ยังไม่มีการศึกษาชัดเจน
- ใช้ในการตรวจติดตามกรณีที่พบความผิดปกติทางโครโมโซมจากการตรวจ CMA หรือ ใช้ในการประเมินพันธุกรรมของพ่อแม่ผู้ป่วย
Reference
- Practice Committee of the Society for Assisted Reproductive Technology, Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril 2007; 88:1497.
- Verlinsky Y, Rechitsky S, Verlinsky O, et al. Prepregnancy testing for single-gene disorders by polar body analysis. Genet Test 1999; 3:185.
- Verlinsky Y, Rechitsky S, Verlinsky O, et al. Preimplantation diagnosis for sonic hedgehog mutation causing familial holoprosencephaly. N Engl J Med 2003; 348:1449.
- Verlinsky Y, Rechitsky S, Cieslak J, et al. Preimplantation diagnosis of single gene disorders by two-step oocyte genetic analysis using first and second polar body. Biochem Mol Med 1997; 62:182.
- Carson SA, Gentry WL, Smith AL, Buster JE. Trophectoderm microbiopsy in murine blastocysts: comparison of four methods. J Assist Reprod Genet 1993; 10:427.
- Sermon K, Van Steirteghem A, Liebaers I. Preimplantation genetic diagnosis. Lancet 2004; 363:1633.
- Veiga A, Sandalinas M, Benkhalifa M, et al. Laser blastocyst biopsy for preimplantation diagnosis in the human. Zygote 1997; 5:351.
- wendy A. Karyotype Analysis andChromosome Banding. ENCYCLOPEDIA OF LIFE SCIENCES, Nature Publishing Group,2001
- Joyce C, Gary H. The use of arrays in preimplantation genetic diagnosis and screening. Fertility and sterility 2010;94:1173-1177
- ACOG Committee Opinion No. 446: array comparative genomic hybridization in prenatal diagnosis. Obstet Gynecol 2009; 114:1161.
- Manning M, Hudgins L, Professional Practice and Guidelines Committee. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med 2010; 12:742.
- Hillman S, Prtelove S, Coomarasamy A, et al. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstet Gynecol 2011; 37: 6–14
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012; 367:2175.
- Anthony J, June C, Konstantina H. Comparative Genomic Hybridization–Array Analysis Enhances the Detection of Aneuploidies and Submicroscopic Imbalances in Spontaneous Miscarriages. Am. J. Hum. Genet 2004; 74:1168–1174
- Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 1991; 49:995.
- Savage MS, Mourad MJ, Wapner RJ. Evolving applications of microarray analysis in prenatal diagnosis. Curr Opin Obstet Gynecol 2011; 23:103.
- Breman A, Pursley AN, Hixson P, et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat Diagn 2012; 32:351.
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev 2007;17:182–92.
- Chung M, Jeong H, et al.. Comprehensive chromosome analysis of blastocysts before implantation using array CGH. Molecular Cytogenetics 2013, 6:22
