Disorders of sexual development (DSD)
พญ. ณัฐนิภา ภารพบ
อาจารย์ที่ปรึกษา ผศ.พญ. อุบล แสงอนันต์
ลักษณะทางเพศเป็นผลจากการพัฒนาทางเพศที่แตกต่างกันของเพศชายและเพศหญิง ภายใต้การความคุมของยีนและฮอร์โมนต่างๆ การพัฒนาทางเพศประกอบด้วย การกำหนดเพศตามโครโมโซมตั้งแต่มีการปฏิสนธิ การพัฒนาของอวัยวะระบบสืบพันธุ์ซึ่งประกอบด้วยอวัยวะสืบพันธุ์ภายใน(internal reproductive organs) และอวัยวะสืบพันธุ์ภายนอก(external reproductive organs)
ในระยะแรกตั้งแต่มีการปฏิสนธิจนอายุครรภ์7สัปดาห์ ตัวอ่อนทั้งเพศชายและหญิงจะมีลักษณะที่เหมือนกัน ไม่สามารถแยกลักษณะทางเพศได้ เรียกว่า indifferent state อวัยวะสืบพันธุ์ภายในของทั้งสองเพศได้แก่ mesonephric (Wolffian) duct และ paramesonephric(Mullerian) duct ส่วนอวัยวะสืบพันธุ์ภายนอก ประกอบด้วย genital tubercle, genital groove, urogenital folds และ labioscrotal swelling
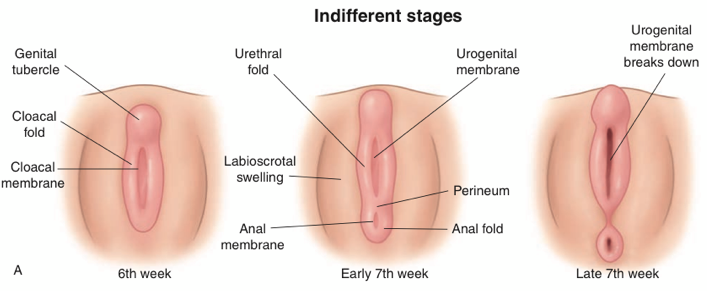
ภาพแสดงการพัฒนาของอวัยวะสืบพันธุ์ภายนอกช่วง undifferentiated state(1)
หลังอายุครรภ์7สัปดาห์ จะเกิดการพัฒนาทางเพศที่แตกต่างกัน เรียกว่า sex differentiation
ตัวอ่อนที่มีโครโมโซมY จะมีSRY gene ซึ่งอยู่บนแขนสั้นของโครโมโซมY สร้าง testis determining factor(TDF) ทำให้มีการพัฒนาของต่อมบ่งเพศ(gonad)ไปเป็นอัณฑะ (2)ภายในอัณฑะประกอบด้วยเซลล์ที่มีผลต่อการพัฒนาทางเพศได้แก่
- Leydig cell สร้าง testosterone ทำให้ mesonephric (Wolffian) duct มีการพัฒนาต่อเป็น epididymis, vas deferens และ seminal vesicle นอกจากนี้ testosterone ยังเปลี่ยนไปเป็น dihydrotestosterone ผ่านเอนไซม์ 5-reductase ซึ่งทำให้เกิดการพัฒนาของอวัยวะสืบพันธุ์ภายนอกเพศชายเป็น penis และ scrotum
- Sertoli cell สร้าง anti-Mullerian hormone (AMH) มีผลยับยั้งการพัฒนาของ paramesonephric (Mullerian) duct
ในเพศหญิง ไม่มีโครโมโซมY ต่อมเพศจึงพัฒนาต่อเป็นรังไข่ ไม่มี anti-Mullerian hormone (AMH) จึงทำให้ paramesonephric (Mullerian) duct เจริญต่อเป็นมดลูก ท่อนำไข่และช่องคลอดส่วนบน นอกจากนี้การที่เพศหญิงไม่มีฮอร์โมน testosterone ทำให้ mesonephric (Wolffian) duct สลายไป(3)
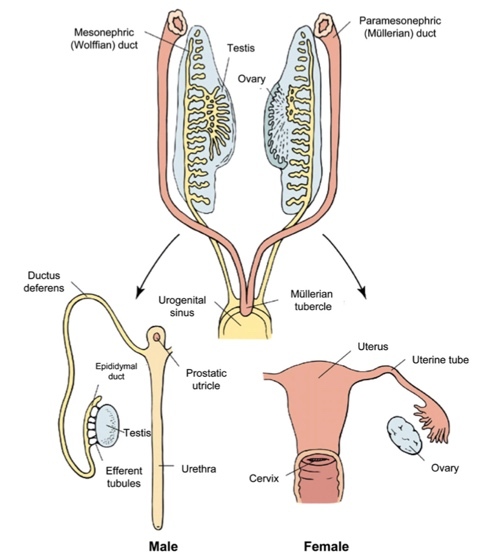
ภาพแสดงการพัฒนาของอวัยวะสืบพันธุ์ภายในจากช่วง undifferentiated state สู่ช่วง sex differentiation(3)
ภาพแสดงการพัฒนาของอวัยวะสืบพันธุ์ภายนอกช่วง sex differentiation(1)
ความผิดปกติในการพัฒนาทางเพศเกิดได้ในทุกขั้นตอนของการพัฒนาทางเพศ ไม่ว่าจะเป็นขั้นตอนการกำหนดเพศโดยโครโมโซม(chromosomal development) การพัฒนาของต่อมเพศ(gonadal development) หรือการพัฒนาอวัยวะสืบพันธุ์(genital development) หากมีการพัฒนาที่ไม่สัมพันธ์กันของขั้นตอนเหล่านี้ จะทำให้มีการพัฒนาทางเพศที่ผิดปกติเกิดขึ้น เรียกว่า Disorders of sexual development(DSD)(4)
ในปัจจุบันมีการเปลี่ยนแปลงคำศัพท์ที่ใช้เรียกความผิดปกติในการพัฒนาการทางเพศ ดังนี้(5)
|
ศัพท์เก่า
|
ศัพท์ปัจจุบัน
|
| Intersex
Male pseudohermaphrodite
Undervirilization of an XY male
Undermasculinizatoin of an XY male
Female pseudohermaphrodite
Overvirilization of an XX female
Masculinizatoin of an XX female
True hermaphrodite
XX male or XX sex reversal
XY sex reversal |
Disorders of sexual development (DSD)
46, XY DSD
46, XX DSD
Ovotesticular DSD
46, XX testicular DSD
46, XY complete gonadal dysgenesis |
Disorder of sexual development (DSD)
แบ่งได้เป็น 3 ประเภท ได้แก่ 46, XY DSD, 46, XX DSD และ Sex chromosome DSD แต่ละประเภทมีโรคที่พบได้ดังแสดงในตาราง(6)
|
Classification
|
Disorders
|
| 46, XY DSD |
- 1.Disorders of testicular development
Complete gonadal dysgenesis (Swyer Syndrome)
Partial gonadal dysgenesis
Gonadal regression
Ovotesticular DSD
- 2.Disorders of androgen synthesis/action
Synthesis: 17-hydroxysteroid dehydrogenase หรือ 5a-reductase deficiency
Action: complete or partial androgen insensitivity syndromes
Receptor defect: Leydig cell hypoplasia
Disorders of AMH and receptor: persistent Mullerian duct syndrome
- 3.Others: Severe hypospadias, Cloacal exstrophy
|
| 46, XX DSD |
- 1.Disorders of ovarian development
Ovotesticular DSD
Testicular DSD (เช่น duplication SOX9)
Gonadal dysgenesis
- 2.Androgen excess
Fetal: congenital adrenal hyperplasia (21-hydroxylase deficiency,
11-hydroxylase deficiency)
Fetoplacental: aromatase deficiency
Maternal: luteoma, exogenous
|
| Sex chromosome DSD |
45, XO (Turner’s syndrome)
47, XXY (Klinefelter’s syndrome and variants)
45, XO/46, XY (mixed gonadal dysgenesis, ovotesticular DSD)
46, XX/46, XY (chimeric, ovotesticular DSD) |
โรคที่พบบ่อยในกลุ่ม 46, XY DSD ได้แก่ Swyer syndrome (complete gonadal dysgenesis), 5-reductase deficiency และ Androgen insensitivity syndromes(AIS)
Swyer syndrome
Swyer syndrome เกิดจากความผิดปกติในการพัฒนาของต่อมเพศโดยสมบูรณ์ (complete gonadal dysgenesis) โดยมีอุบัติการณ์ 1:80,000 ของการคลอดมีชีพทั้งหมด เชื่อว่า10-20%เกิดจากการmutationของSRY gene ทำให้ต่อมเพศไม่มีการพัฒนาไปเป็นอัณฑะ ต่อมเพศฝ่อ มีลักษณะเป็นstreak gonad ไม่สามารถสร้างtestosteroneได้(7)
ลักษณะอวัยวะสืบพันธุ์ทั้งภายนอกและภายในเป็นเพศหญิง เมื่อเข้าสู่วัยเข้าสู่วัยรุ่น จะไม่มีการพัฒนาทางเพศขั้นที่สอง ผู้ป่วยมักมาพบแพทย์ด้วยปัญหา primary amenorrhea
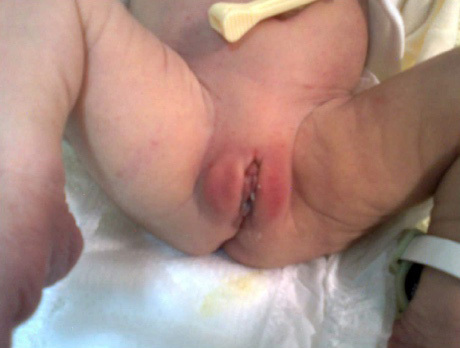
ภาพแสดงลักษณะอวัยวะเพศภายนอกผู้ป่วย Swyer syndrome(8)
การวินิจฉัย อาการแสดงที่เข้าได้ การตรวจโครโมโซม และพบระดับฮอร์โมนจากต่อมใต้สมอง (FSH,LH) สูง
การรักษา
- ฮอร์โมนestrogenและprogesteroneเสริม เพื่อกระตุ้นให้มีการพัฒนาการทางเพศขั้นที่สองและป้องกันภาวะendometrial hyperplasia ตามลำดับ
- Gonadectomy ทันทีที่ได้รับการวินิจฉัย เนื่องจากมีโอกาสที่จะกลายไปเป็นมะเร็งได้สูง
5-reductase deficiency
5-reductase deficiency เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบ autosomal recessive โดยเอนไซม์ 5-reductase ทำหน้าที่ในการเปลี่ยนฮอร์โมนtestosteroneไปเป็นdihydrotestosterone (DHT) ซึ่งเป็นฮอร์โมนที่มีผลต่อการพัฒนาลักษณะอวัยวะสืบพันธุ์ภายนอกของเพศชาย
แรกเกิดผู้ป่วยจะมีอวัยวะเพศภายนอกกำกวม ลักษณะดูคล้ายเพศหญิง โดยต่อมเพศเป็นอัณฑะ ท่อสืบพันธุ์ภายในเป็นเพศชาย เมื่อเข้าสู่วัยผู้ใหญ่จะมีลักษณะที่เป็นเพศชายมากขึ้น เนื่องจากมีระดับtestosteroneสะสมสูงขึ้น
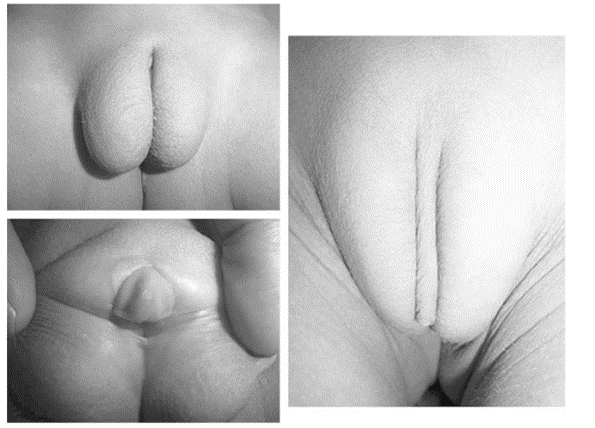
ภาพแสดงลักษณะอวัยวะเพศผู้ป่วย 5-reductase deficiency(9)
การวินิจฉัย อาการแสดงที่เข้าได้ ระดับtestosteroneที่สูง ระดับ DHTที่ต่ำ และค่าtestosterone/DHT>20 (10)
การรักษา ขึ้นอยู่กับลักษณะการแสดงออกทางเพศ เวลาที่วินิจฉัย
- กรณีที่ได้รับการเลี้ยงดูเป็นเพศหญิง ควรได้รับการผ่าตัด gonadectomy ก่อนเข้าสู่ช่วงวัยรุ่น และให้estrogenเสริมเมื่อเริ่มเข้าสู่วัยรุ่นหรือทันทีหลังผ่าตัด เพื่อคงความเป็นเพศหญิง
- กรณีที่ได้รับการเลี้ยงดูเป็นเพศชาย พิจารณาผ่าตัดหากมีภาวะhypospadiasและcryptorchidism
Androgen insensitivity syndrome (AIS)
Androgen insensitivity syndrome (AIS) เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบ X-linked recessive เกิดจากการmutationของ androgen receptor gene ทำให้เกิดความผิดปกติในการออกฤทธิ์ของandrogen แบ่งได้เป็น 2 กลุ่มตามความผิดปกติในการออกฤทธิ์ของandrogen คือ complete androgen insensitivity syndrome (CAIS) และ partial androgen insensitivity syndrome (PAIS) (11)
ผู้ป่วยที่เป็น complete androgen insensitivity syndrome (CAIS) จะมีลักษณะอวัยวะสืบพันธุ์ภายนอกเป็นเพศหญิงโดยสมบูรณ์ มีพัฒนาการทางเพศขั้นที่สองไม่ดี ไม่มีขนรักแร้และขนหัวหน่าวหรืออาจจะมีน้อย ช่องคลอดเป็นแอ่งตื้น มักมาพบแพทย์ด้วยปัญหา primary amenorrhea หรือปัญหาคลำได้ก้อนบริเวณท้อง มีไส้เลื่อนบริเวณขาหนีบซึ่งเกิดจากต่อมเพศ(อัณฑะ) ไม่เลื่อนลงถุงอัณฑะ
การวินิจฉัย การตรวจโครโมโซม การตรวจsequence analysisของandrogen receptor
ผู้ป่วยที่เป็น partial androgen insensitivity syndrome (PAIS) มีการแสดงออกได้ในหลายลักษณะ ตั้งแต่เกือบเป็นหญิงโดยสมบูรณ์จนเกือบเป็นชายโดยสมบูรณ์ ขึ้นอยู่กับระดับความรุนแรงของโรค
การวินิจฉัย การตรวจโครโมโซม และระดับ androstenedione, testosterone, DHT
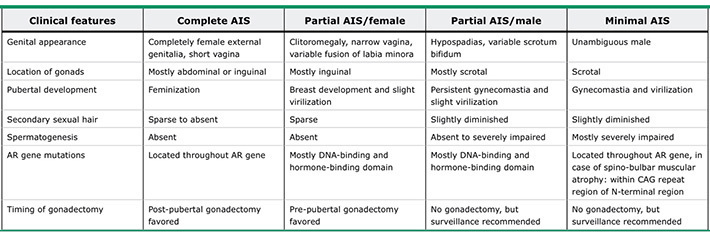
ตารางเปรียบเทียบสาเหตุ การพัฒนาทางเพศ ลักษณะทางเพศ และช่วงเวลาในการพิจารณาการรักษาของผู้ป่วยกลุ่มAIS
การรักษา
- ให้คำแนะนำ ดูแลสภาจิตใจผู้ป่วยและครอบครัว
- ประเมินเพศสภาพของผู้ป่วย
- พิจารณา gonadectomy เพื่อป้องกันการเกิดเนื้องอกของต่อมเพศ ซึ่งช่วงเวลาในการพิจารณาผ่าตัดจะแตกต่างกันตามความรุนแรงของโรค
- ให้ฮอร์โมนestrogenเสริม ในผู้ป่วยที่ได้รับการเลี้ยงดูเป็นเพศหญิง
- ให้ฮอร์โมนtestosteroneเสริม ในผู้ป่วยที่ได้รับการเลี้ยงดูเป็นเพศชาย
โรคที่พบบ่อยในกลุ่ม 46, XX DSD ได้แก่ Congenital adrenal hyperplasia (CAH)
Congenital adrenal hyperplasia (CAH)
Congenital adrenal hyperplasia (CAH) เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบ autosomal recessive
มีอุบัติการณ์ 1:15,000 ของการคลอดมีชีพทั้งหมด เป็นสาเหตุในการเกิดอวัยวะเพศกำกวมที่พบได้บ่อยที่สุด เกิดจากการขาดเอนไซม์ในการสร้างฮอร์โมนจากต่อมหมวกไต ทำให้ระดับcortisolลดลง ร่างกายจึงเกิดการตอบสนองโดยการสร้างACTHจากต่อมใต้สมองมากขึ้น ผลจากACTHที่มากขึ้นส่งผลให้เกิดการกระตุ้นต่อมหมวกไต จนเกิดเป็น adrenal hyperplasia
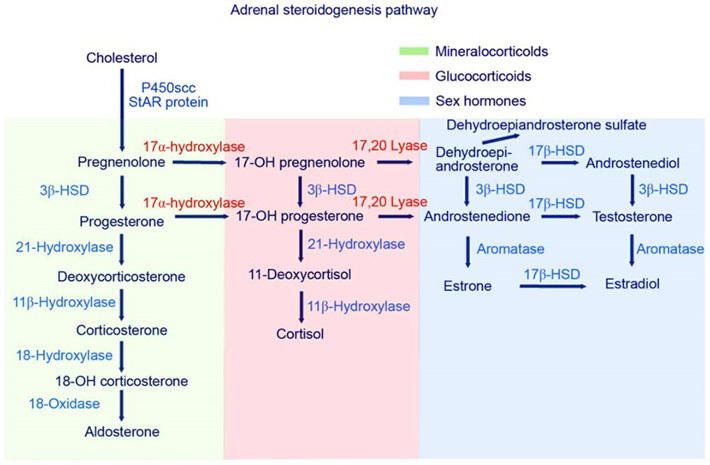
แผนภาพแดงการสร้าง steroid hormone จากadrenal cortex(12)
สาเหตุของ CAH เกิดได้จากการขาดเอนไซม์ 21-hydroxylase, 11b-hydroxylase, 3β-hydroxysteroid dehydrogenase โดยภาวะ 21-hydroxylase deficiency เป็นสาเหตุที่พบบ่อยที่สุดและเป็นสาเหตุที่ทำให้ทารกเสียชีวิตจากระบบต่อมไร้ท่อมากที่สุด
ภาวะ 21-hydroxylase deficiency แบ่งได้เป็น3ชนิดตามระดับความรุนแรง ได้แก่
- Salt wasting พบได้ 75% ผู้ป่วยจะขาดcortisolและaldosteroneรุนแรง หากไม่ได้รับการรักษาจะทำให้ขาดน้ำ ระดับโซเดียมต่ำ โพแทสเซียมสูง จนเกิดadrenal crisisได้ในช่วง1-4สัปดาห์หลังคลอดและจะเสียชีวิตได้หากไม่ได้รับฮอร์โมนทดแทน ทารกมีลักษณะอวัยวะเพศกำกวม
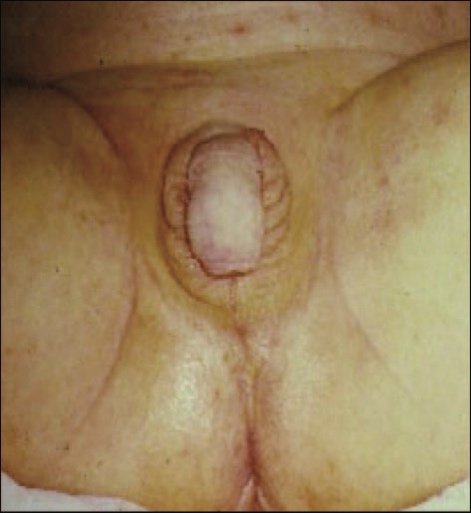
ภาพแสดงลักษณะอวัยวะเพศผู้ป่วย 21-hydroxylase deficiency (salt wasting form)(13)
- Simple virilizing พบได้ 25% ผู้ป่วยจะมีการทำงานของ 21-hydroxylase ที่มากกว่าชนิดแรก มีความรุนแรงของโรคน้อยกว่า สามารถสร้างaldosteroneได้เพียงพอที่จะไม่ทำให้เกิดภาวะadrenal crisis แต่ยังมีระดับandrogenสูง จึงยังพบลักษณะความเป็นเพศชายได้
- Non classical form พบได้น้อยและระดับความรุนแรงของโรคน้อยที่สุดในกลุ่ม 21-hydroxylase deficiency เนื่องจากยังมีcortisolและaldosterone แต่มีในระดับต่ำกว่าคนปกติ ยังมีอาการแสดงจากภาวะhyperandrogenได้ เช่น มีขนที่หัวหน่าวก่อนวัย สิวขึ้น ขนดก มีบุตรยาก
การวินิจฉัย ตรวจระดับ morning 17-OHP มีค่าสูง >800-1,000 ng/dL (>24-30 nmol/L) ซึ่งค่าปกติ <200ng/dL (<6 nmol/L) ในผู้ป่วยที่มีค่า morning 17-OHP ก่ำกึ่ง (200-800 nmol/L) จะทำการตรวจ cosyntropin(ACTH) stimulation test ยืนยันการวินิจฉัย ซึ่งแปลผลได้ดังแสดงในกราฟ
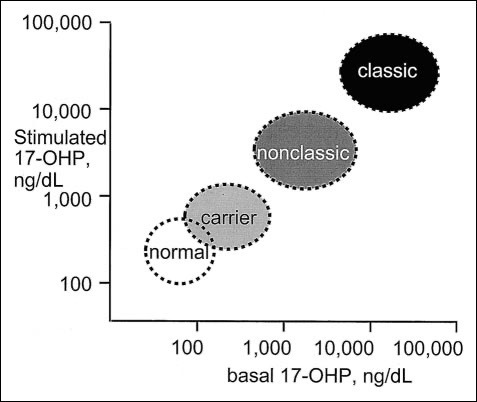
แสดงการแปลผลการตรวจ cosyntropin(ACTH) stimulation test(13)
การรักษา
- ป้องกันการเกิดภาวะ adrenal crisis และvirilization (เป้าหมายหลักในการรักษา)
- Hydrocortisone, mineralocorticoid, sodium chloride supplement
- การผ่าตัดรักษาอวัยวะเพศกำกวม โดยทำclitoroplastyและvaginoplastyเป็นหลัก
โรคที่พบบ่อยในกลุ่ม Chromosomal DSD ได้แก่ Klinefelter’s syndrome และ Turner’s syndrome
Klinefelter’s syndrome
Klinefelter’s Syndrome เป็นโรคที่เกิดจากความผิดปกติของโครโมโซมเพศในช่วงgamatogenesis มีโครโมโซมเป็น 46, XXY มีอุบัติการณ์ 1:1,000 ของการคลอดมีชีพทั้งหมด
ทารกที่เป็น Klinefelter’s Syndrome จะมีลักษณะคล้ายทารกปกติ แต่อาจพบอวัยวะเพศมีขนาดเล็ก(micropenis), รูเปิดท่อปัสสาวะต่ำกว่าปกติ(hypospadias) หรืออัณฑะไม่เลื่อนลงถุง(cryptorchidism)ได้ ผู้ป่วยส่วนใหญ่จะได้รับการวินิจฉัยเมื่อเข้าสู่วัยผู้ใหญ่ ซึ่งมักมาพบแพทย์ด้วยภาวะมีบุตรยาก สาเหตุเกิดจากการที่ต่อมเพศไม่เจริญ(gonadal dysgenesis) นอกจากนี้ยังมีลักษณะอื่นๆที่พบได้บ่อย ได้แก่ ตัวสูง หน้าอกใหญ่(gynecomastia) อวัยวะเพศและอัณฑะมีขนาดเล็ก มีขนตามร่างกายน้อย ความต้องการทางเพศน้อย กระดูกพรุน มีพัฒนาการทางสติปัญญาต่ำกว่าเกณฑ์

ภาพแสดงลักษณะผู้ป่วย Klinefelter’s Syndrome(14)
การวินิจฉัย การตรวจโครโมโซม
การรักษา
- ให้ฮอร์โมนtestosteroneเสริม
- ผ่าตัดลดขนาดเต้านม
Turner’s syndrome
Turner’s syndrome เป็นความผิดปกติของโครโมโซมเพศที่เกิดในเพศหญิง โดยโครโมโซมX หายไปบางส่วนหรือทั้งหมด มีอุบัติการณ์ 1:2,000 ของการคลอดมีชีพทั้งหมด 45%ของทารกที่มีชีวิตรอดจะมีลักษณะโครโมโซมเป็น 45, XO (monosomy X) 50%จะเป็น 45, X0 with mosaicism เช่น 45,X0/46,XX หรือ 45,X0/46,XY
ลักษณะที่พบได้แก่ ตัวเตี้ย, มีภาวะovarian failure(ไม่เข้าสู่วัยสาว ไม่มีประจำเดือน เป็นหมัน), มีแผงคอ(webbed neck), ลักษณะทางเพศภายนอกเป็นเพศหญิงปกติ นอกจากนี้อาจมีความผิดปกติของระบบอื่นๆในรางกายร่วมด้วย
ภาพแสดงลักษณะผู้ป่วย Turner’s syndrome(15)
ตารางแสดงความผิดปกติที่พบได้ในระบบต่างๆของร่างกาย(16)
|
ระบบร่างกาย
|
ตัวอย่างความผิดปกติที่พบบ่อย
|
| Skeletal disturbance |
Short stature, growth failure, increase upper to lower segment ratio, defect in dental development, cubitus valgus |
| Lymphatic obstruction
|
Low posterior hairline, edema of hands/feet, webbed neck |
| Germ cell defects
|
Infertility, ovarian failure,gonadal dysgenesis |
| Cardiac malformation
|
Elongated transverse aortic arch, aortic valve malformation, coarctation of aorta |
| Renal anomalies
|
Collecting system malformation, horseshoe kidney |
| Ocular abnormalities
|
Myopia/hypopia, strabismus, ptosis |
| Ears and hearing
|
Recurrent otitis media, SNHL, CHL |
| Autoimmune
|
Thyroiditis |
การวินิจฉัย การตรวจโครโมโซม ตรวจหาความผิดปกติของระบบต่างๆในร่างกายเพิ่มเติม
การรักษา
- ให้growth hormoneและestrogenเสริม
- รักษาความผิดปกติของระบบอื่นๆที่พบ
การดูแลผู้ป่วยที่มีความผิดปกติของการพัฒนาทางเพศ อาศัยการซักประวัติ ตรวจร่างกายโดยละเอียด การส่งตรวจเพิ่มเติมอย่างเหมาะสม และอาศัยสหสาขาวิชาชีพในการดูแลผู้ป่วย ไม่ว่าจะเป็นกุมารแพทย์ สูตินรีแพทย์ จิตแพทย์ และศัลยแพทย์ ร่วมกันวางแผนการรักษาที่ถูกต้องและเหมาะสม
เอกสารอ้างอิง
- FG C. Williams obstetrics. 25th edition ed. New York: McGraw-Hill; 2018.
- Witchel SF. Disorders of sex development. Best Practice & Research Clinical Obstetrics & Gynaecology. 2018;48:90-102.
- Biason-Lauber A. Control of sex development. Best practice & research Clinical endocrinology & metabolism. 2010;24(2):163-86.
- Öçal G. Current concepts in disorders of sexual development. Journal of Clinical Research in Pediatric Endocrinology. 2011;3(3):105.
- Hughes IA. Disorders of sex development: a new definition and classification. Best practice & research Clinical endocrinology & metabolism. 2008;22(1):119-34.
- Woodward M, Burns K. Disorders of sex development. Surgery (Oxford). 2019;37(11):646-52.
- Iliopoulos D, Volakakis N, Tsiga A, Rousso I, Voyiatzis N, editors. Description and molecular analysis of SRY and AR genes in a patient with 46, XY pure gonadal dysgenesis (Swyer syndrome). Annales de génétique; 2004: Elsevier.
- Machado CL, Pereira nGe, Cruz JM, Cadilhe A, Silva A, Pereira A, editors. A novel SRY nonsense mutation in a case of Swyer syndrome2014.
- Sahakitrungruang T, Wacharasindhu S, Yeetong P, Snabboon T, Suphapeetiporn K, Shotelersuk V. Identification of mutations in the SRD5A2 gene in Thai patients with male pseudohermaphroditism. Fertility and sterility. 2008;90(5):2015. e11-. e15.
- Peterson RE, Imperato-McGinley J, Gautier T, Sturla E. Male pseudohermaphroditism due to steroid 5α-reductase deficiency. The American journal of medicine. 1977;62(2):170-91.
- Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. The Lancet. 2012;380(9851):1419-28.
- Xu S, Hu S, Yu X, Zhang M, Yang Y. 17α‑hydroxylase/17, 20‑lyase deficiency in congenital adrenal hyperplasia: A case report. Molecular medicine reports. 2017;15(1):339-44.
- Antal Z, Zhou P. Congenital Adrenal Hyperplasia. Pediatrics in review. 2009;30(7):e49.
- Bojesen AB, H¯st C, Gravholt CHj. Klinefelter’s syndrome, type 2 diabetes and the metabolic syndrome: the impact of body composition. Molecular human reproduction. 2010;16 6:396-401.
- Lui B. Turner syndrome. 1988.
- Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome. European journal of endocrinology. 2004;151(6):657-88.